

Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1
- PMID: 25818337
- PMCID: PMC4422247
- DOI: 10.2215/CJN.10141014
Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1
Abstract
Background and objectives: The Wilms tumor suppressor gene 1 (WT1) plays an essential role in urogenital and kidney development. Genotype/phenotype correlations of WT1 mutations with renal function and proteinuria have been observed in world-wide cohorts with nephrotic syndrome or Wilms tumor (WT). This study analyzed mid-European patients with known constitutional heterozygous mutations in WT1, including patients without proteinuria or WT.
Design, setting, participants & measurements: Retrospective analysis of genotype, phenotype, and treatment of 53 patients with WT1 mutation from all pediatric nephrology centers in Germany, Austria, and Switzerland performed from 2010 to 2012.
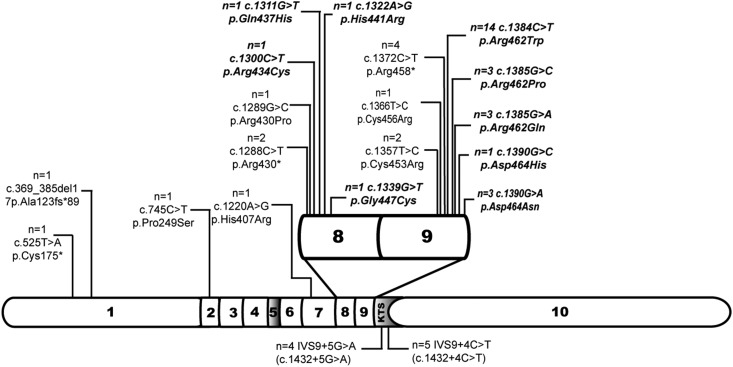
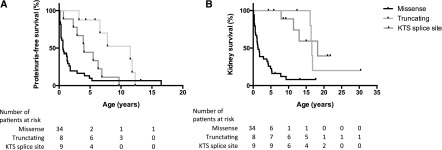
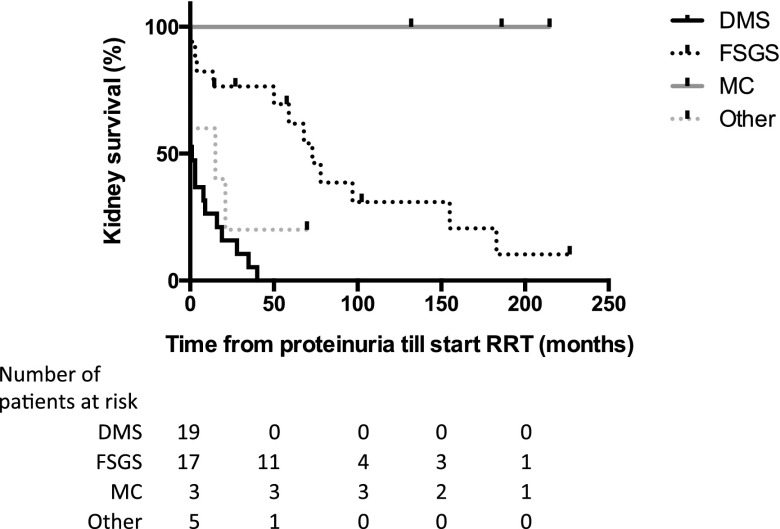
Results: Median age was 12.4 (interquartile range [IQR], 6-19) years. Forty-four of 53 (83%) patients had an exon mutation (36 missense, eight truncating), and nine of 53 (17%) had an intronic lysine-threonine-serine (KTS) splice site mutation. Fifty of 53 patients (94%) had proteinuria, which occurred at an earlier age in patients with missense mutations (0.6 [IQR, 0.1-1.5] years) than in those with truncating (9.7 [IQR, 5.7-11.9]; P<0.001) and splice site (4.0 [IQR, 2.6-6.6]; P=0.004) mutations. Thirteen of 50 (26%) were treated with steroids and remained irresponsive, while three of five partially responded to cyclosporine A. Seventy-three percent of all patients required RRT, those with missense mutations significantly earlier (at 1.1 [IQR, 0.01-9.3] years) than those with truncating mutations (16.5 [IQR, 16.5-16.8]; P<0.001) and splice site mutations (12.3 [IQR, 7.9-18.2]; P=0.002). Diffuse mesangial sclerosis was restricted to patients with missense mutations, while focal segmental sclerosis occurred in all groups. WT occurred only in patients with exon mutations (n=19). Fifty of 53 (94%) patients were karyotyped: Thirty-one (62%) had XY and 19 (38%) had XX chromosomes, and 96% of male karyotypes had urogenital malformations.
Conclusions: Type and location of WT1 mutations have predictive value for the development of proteinuria, renal insufficiency, and WT. XY karyotype was more frequent and associated with urogenital malformations in most cases.
Keywords: genetic renal disease; nephrotic syndrome; pediatric nephrology.
Copyright © 2015 by the American Society of Nephrology.
Figures
References
-
- Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE: Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60: 509–520, 1990 - PubMed
-
- Gessler M, König A, Arden K, Grundy P, Orkin S, Sallan S, Peters C, Ruyle S, Mandell J, Li F, Cavenee W, Bruns G: Infrequent mutation of the WT1 gene in 77 Wilms’ Tumors. Hum Mutat 3: 212–222, 1994 - PubMed
-
- Hohenstein P, Hastie ND: The many facets of the Wilms’ tumour gene, WT1. Hum Mol Genet 15: R196–R201, 2006 - PubMed
-
- Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D, Van Heyningen V, Hastie N: The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 346: 194–197, 1990 - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
