

Matrix metalloproteinase-2 negatively regulates cardiac secreted phospholipase A2 to modulate inflammation and fever
- PMID: 25820137
- PMCID: PMC4579961
- DOI: 10.1161/JAHA.115.001868
Matrix metalloproteinase-2 negatively regulates cardiac secreted phospholipase A2 to modulate inflammation and fever
Abstract
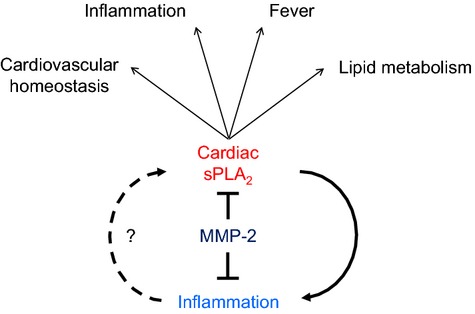
Background: Matrix metalloproteinase (MMP)-2 deficiency makes humans and mice susceptible to inflammation. Here, we reveal an MMP-2-mediated mechanism that modulates the inflammatory response via secretory phospholipase A2 (sPLA2), a phospholipid hydrolase that releases fatty acids, including precursors of eicosanoids.
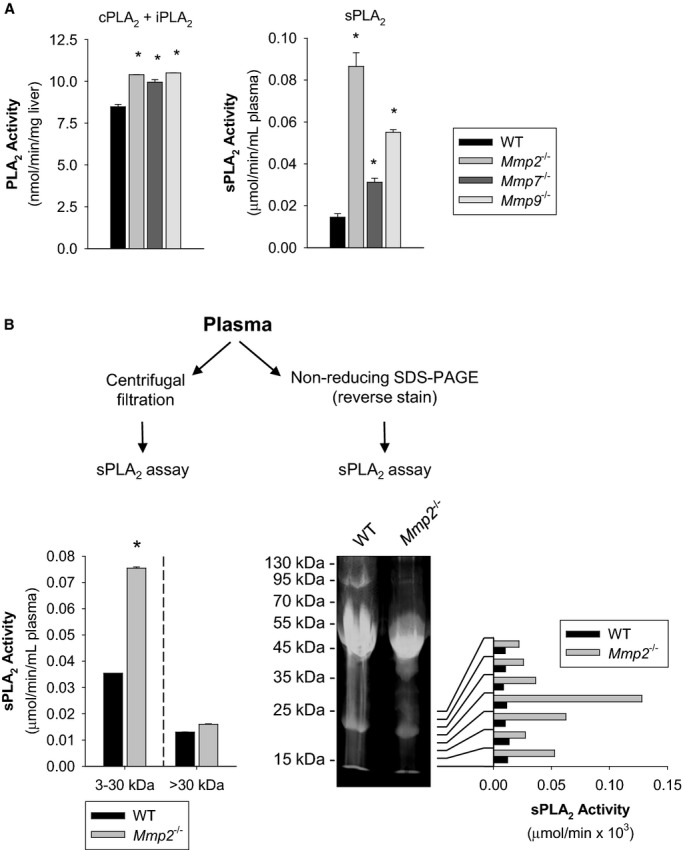
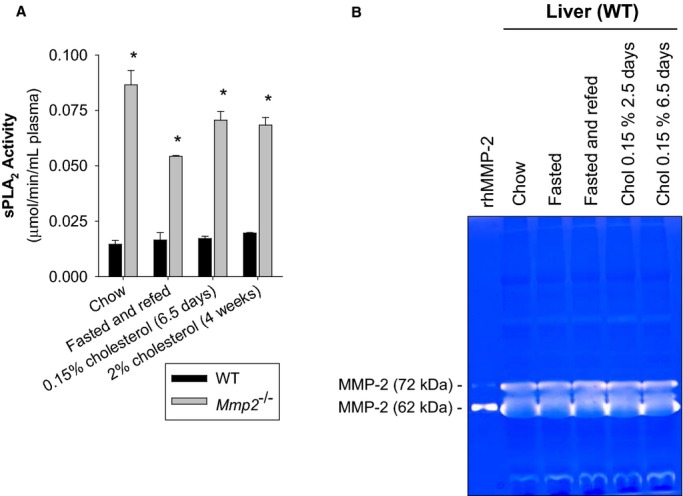
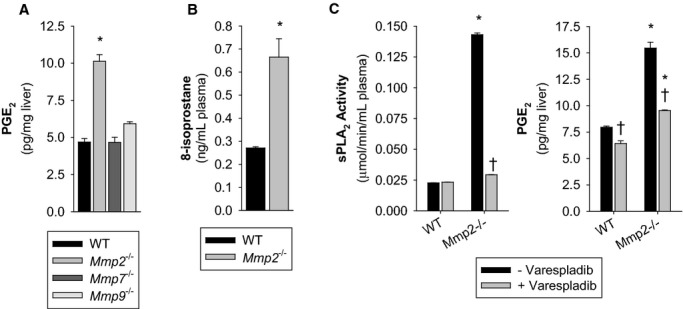
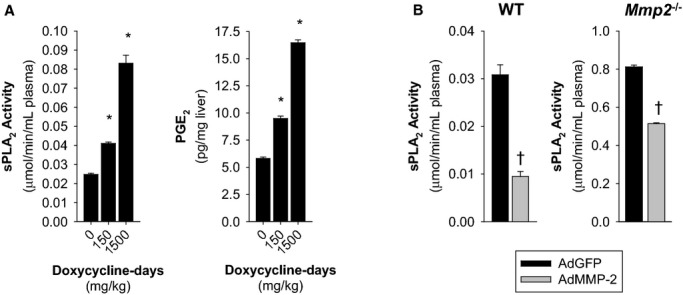
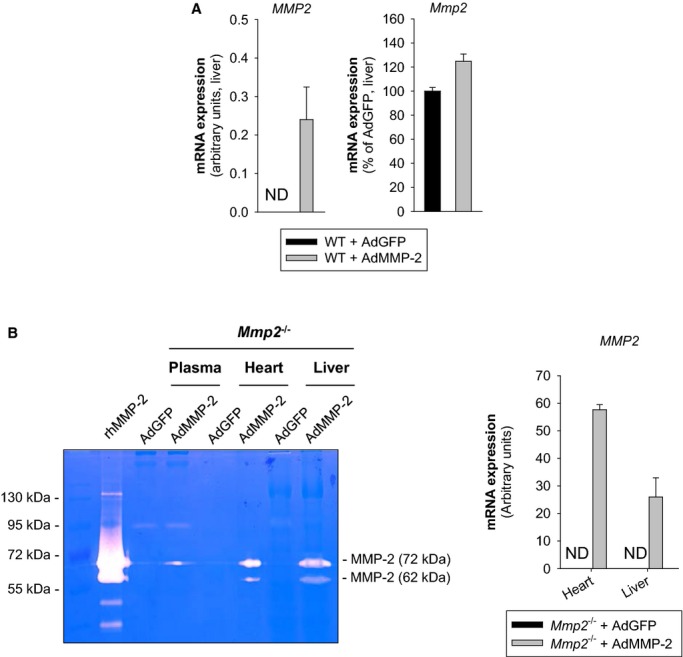
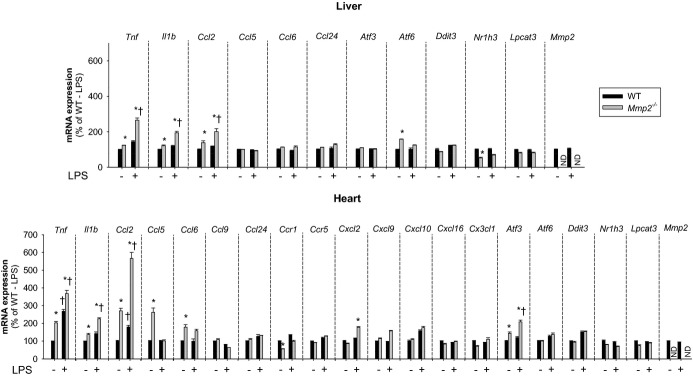
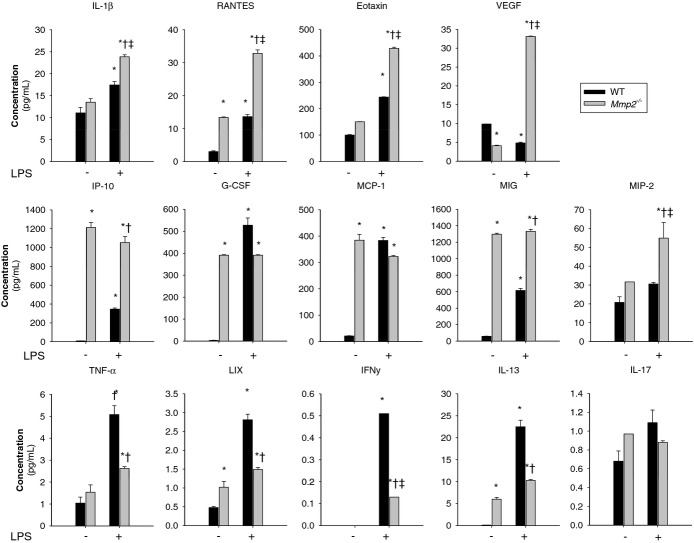
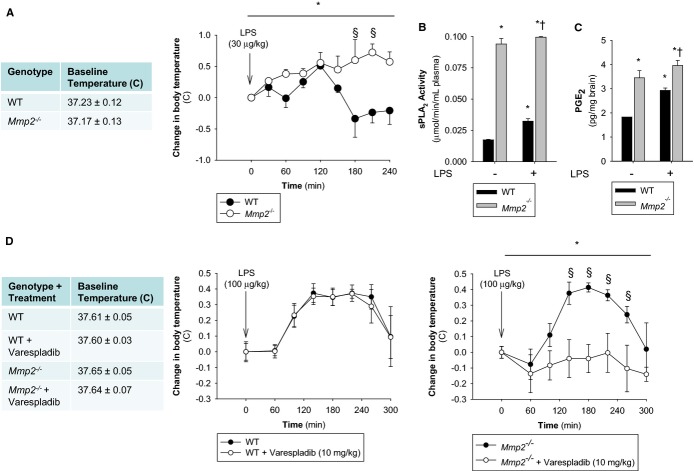
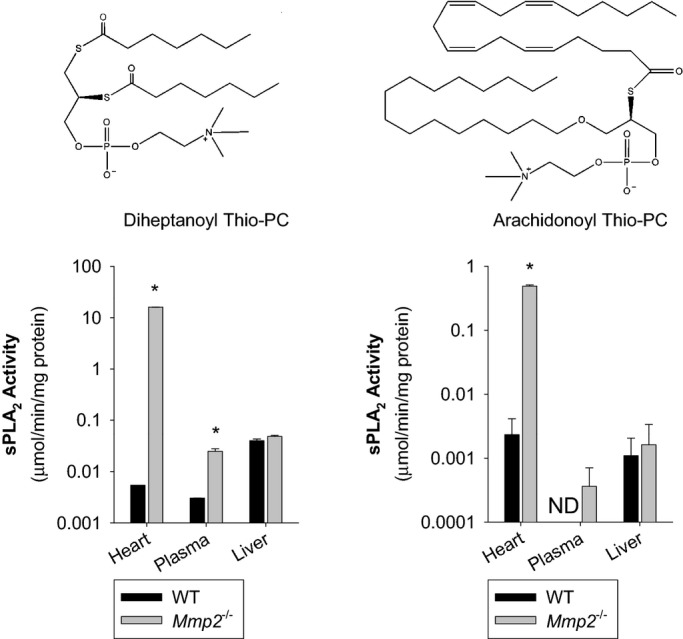
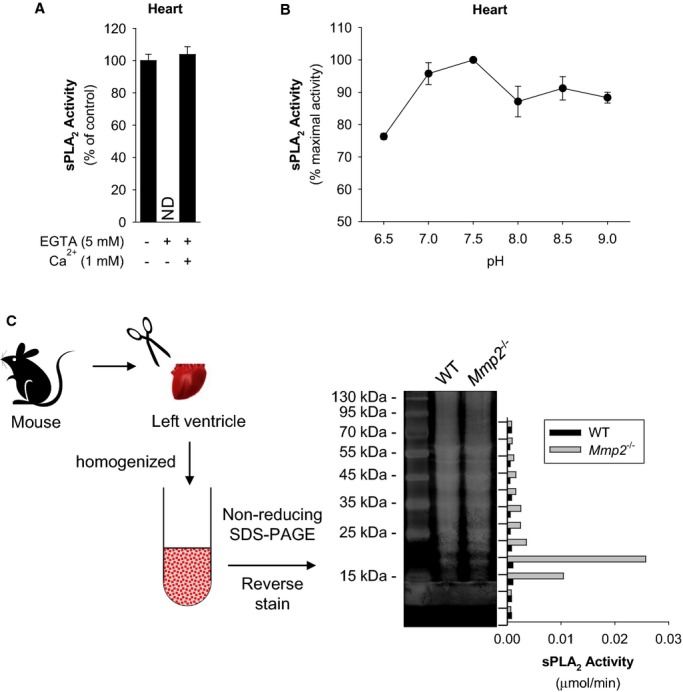
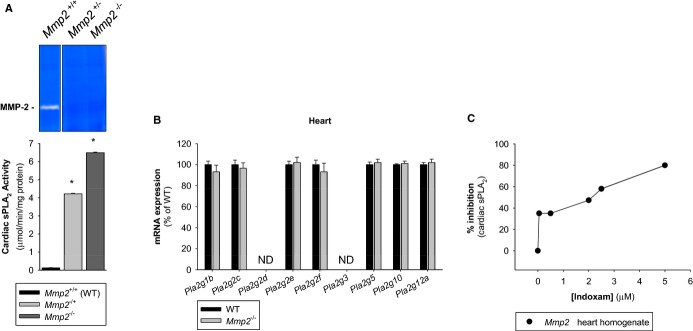
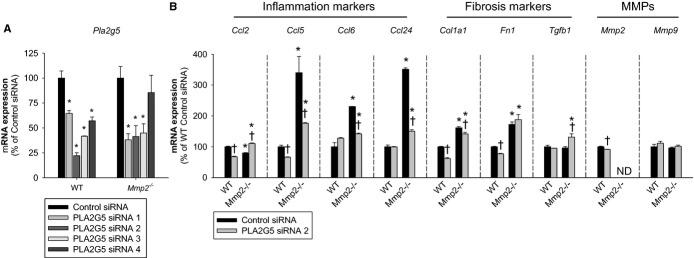
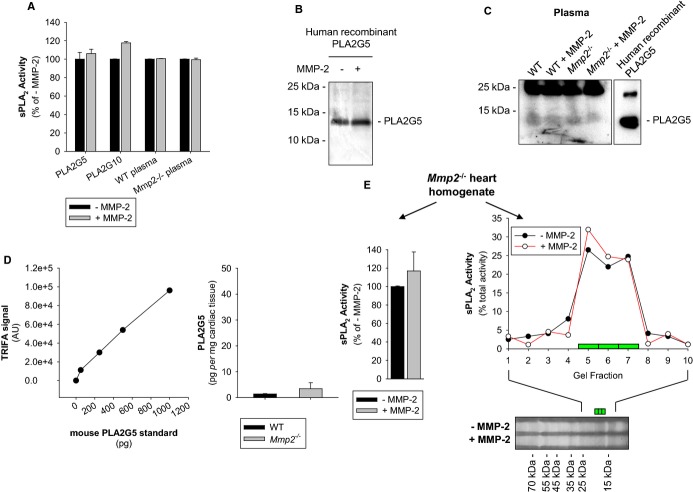
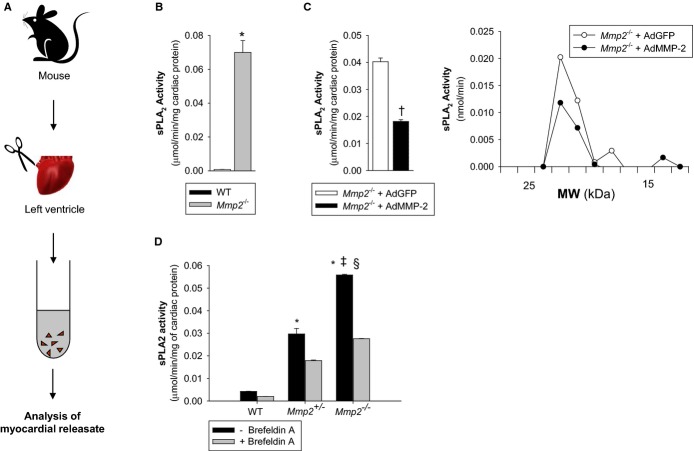
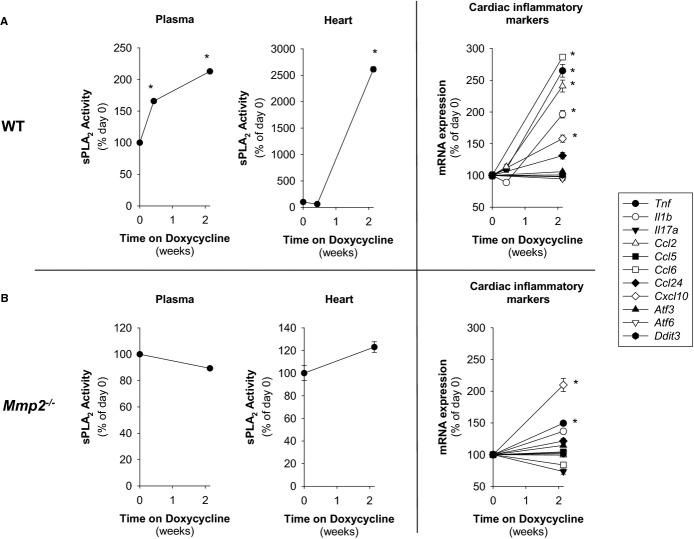
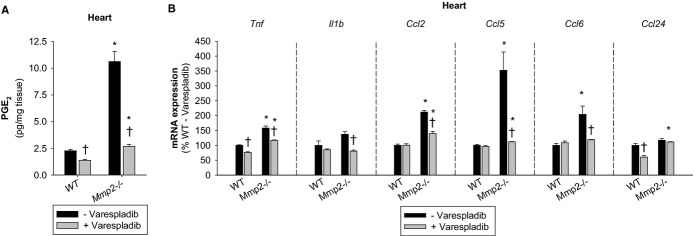
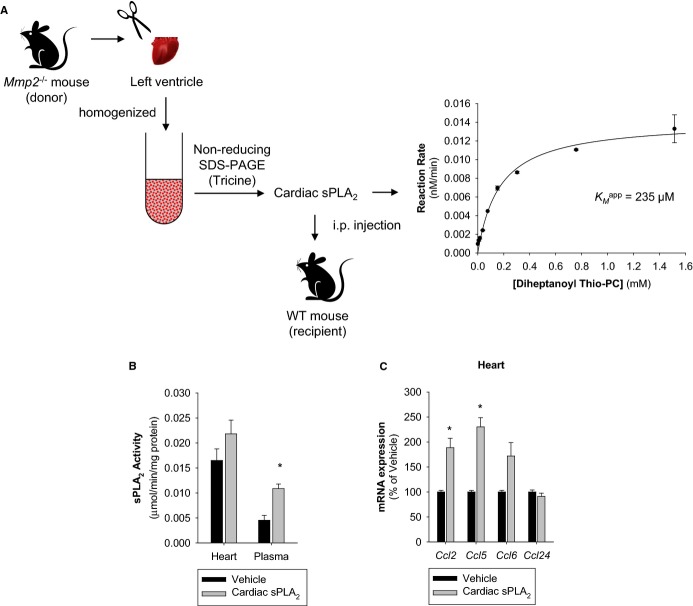
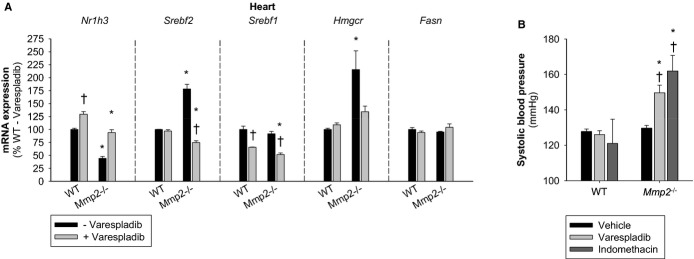
Methods and results: Mmp2(-/-) (and, to a lesser extent, Mmp7(-/-) and Mmp9(-/-)) mice had between 10- and 1000-fold elevated sPLA2 activity in plasma and heart, increased eicosanoids and inflammatory markers (both in the liver and heart), and exacerbated lipopolysaccharide-induced fever, all of which were blunted by adenovirus-mediated MMP-2 overexpression and varespladib (pharmacological sPLA2 inhibitor). Moreover, Mmp2 deficiency caused sPLA2-mediated dysregulation of cardiac lipid metabolic gene expression. Compared with liver, kidney, and skeletal muscle, the heart was the single major source of the Ca(2+)-dependent, ≈20-kDa, varespladib-inhibitable sPLA2 that circulates when MMP-2 is deficient. PLA2G5, which is a major cardiac sPLA2 isoform, was proinflammatory when Mmp2 was deficient. Treatment of wild-type (Mmp2(+/+)) mice with doxycycline (to inhibit MMP-2) recapitulated the Mmp2(-/-) phenotype of increased cardiac sPLA2 activity, prostaglandin E2 levels, and inflammatory gene expression. Treatment with either indomethacin (to inhibit cyclooxygenase-dependent eicosanoid production) or varespladib (which inhibited eicosanoid production) triggered acute hypertension in Mmp2(-/-) mice, revealing their reliance on eicosanoids for blood pressure homeostasis.
Conclusions: A heart-centric MMP-2/sPLA2 axis may modulate blood pressure homeostasis, inflammatory and metabolic gene expression, and the severity of fever. This discovery helps researchers to understand the cardiovascular and systemic effects of MMP-2 inhibitors and suggests a disease mechanism for human MMP-2 gene deficiency.
Keywords: PLA2; heart; inflammation; matrix metalloproteinases.
© 2015 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures
References
-
- Lijnen HR, Collen D. Matrix metalloproteinase system deficiencies and matrix degradation. Thromb Haemost. 1999; 82:837-845. - PubMed
-
- Fraser H, Hislop C, Christie RM, Rick HL, Reidy CA, Chouinard ML, Eacho PI, Gould KE, Trias J. Varespladib (A‐002), a secretory phospholipase A(2) inhibitor, reduces atherosclerosis and aneurysm formation in ApoE(‐/‐) mice. J Cardiovasc Pharmacol. 2009; 53:60-65. - PubMed
-
- Mosig RA, Dowling O, DiFeo A, Ramirez MC, Parker IC, Abe E, Diouri J, Aqeel AA, Wylie JD, Oblander SA, Madri J, Bianco P, Apte SS, Zaidi M, Doty SB, Majeska RJ, Schaffler MB, Martignetti JA. Loss of MMP‐2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007; 16:1113-1123. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
