

Syndromic parkinsonism and dementia associated with OPA1 missense mutations
- PMID: 25820230
- PMCID: PMC5008165
- DOI: 10.1002/ana.24410
Syndromic parkinsonism and dementia associated with OPA1 missense mutations
Abstract
Objective: Mounting evidence links neurodegenerative disorders such as Parkinson disease and Alzheimer disease with mitochondrial dysfunction, and recent emphasis has focused on mitochondrial dynamics and quality control. Mitochondrial dynamics and mtDNA maintenance is another link recently emerged, implicating mutations in the mitochondrial fusion genes OPA1 and MFN2 in the pathogenesis of multisystem syndromes characterized by neurodegeneration and accumulation of mtDNA multiple deletions in postmitotic tissues. Here, we report 2 Italian families affected by dominant chronic progressive external ophthalmoplegia (CPEO) complicated by parkinsonism and dementia.
Methods: Patients were extensively studied by optical coherence tomography (OCT) to assess retinal nerve fibers, and underwent muscle and brain magnetic resonance spectroscopy (MRS), and muscle biopsy and fibroblasts were analyzed. Candidate genes were sequenced, and mtDNA was analyzed for rearrangements.
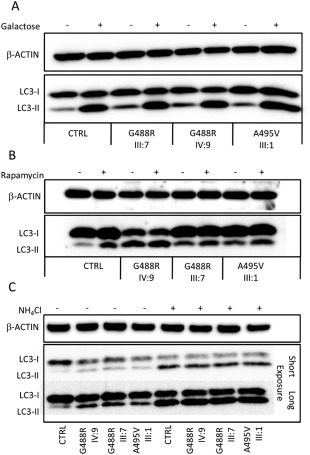
Results: Affected individuals displayed a slowly progressive syndrome characterized by CPEO, mitochondrial myopathy, sensorineural deafness, peripheral neuropathy, parkinsonism, and/or cognitive impairment, in most cases without visual complains, but with subclinical loss of retinal nerve fibers at OCT. Muscle biopsies showed cytochrome c oxidase-negative fibers and mtDNA multiple deletions, and MRS displayed defective oxidative metabolism in muscle and brain. We found 2 heterozygous OPA1 missense mutations affecting highly conserved amino acid positions (p.G488R, p.A495V) in the guanosine triphosphatase domain, each segregating with affected individuals. Fibroblast studies showed a reduced amount of OPA1 protein with normal mRNA expression, fragmented mitochondria, impaired bioenergetics, increased autophagy and mitophagy.
Interpretation: The association of CPEO and parkinsonism/dementia with subclinical optic neuropathy widens the phenotypic spectrum of OPA1 mutations, highlighting the association of defective mitochondrial dynamics, mtDNA multiple deletions, and altered mitophagy with parkinsonism.
© 2015 The Authors Annals of Neurology published by Wiley Periodicals, Inc. on behalf of American Neurological Association.
Figures









References
-
- Zeviani M, Servidei S, Gellera C, et al. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D‐loop region. Nature 1989;339:309–311. - PubMed
-
- Spinazzola A, Zeviani M. Disorders of nuclear‐mitochondrial intergenomic communication. Biosci Rep 2007;27:39–51. - PubMed
-
- Van Goethem G, Dermaut B, Lofgren A, et al. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001;28:211–212. - PubMed
-
- Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4‐like protein localized in mitochondria. Nat Genet 2001;28:223–231. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
