

On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory
- PMID: 25821417
- PMCID: PMC4358633
- DOI: 10.1021/ct4007585
On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory
Abstract
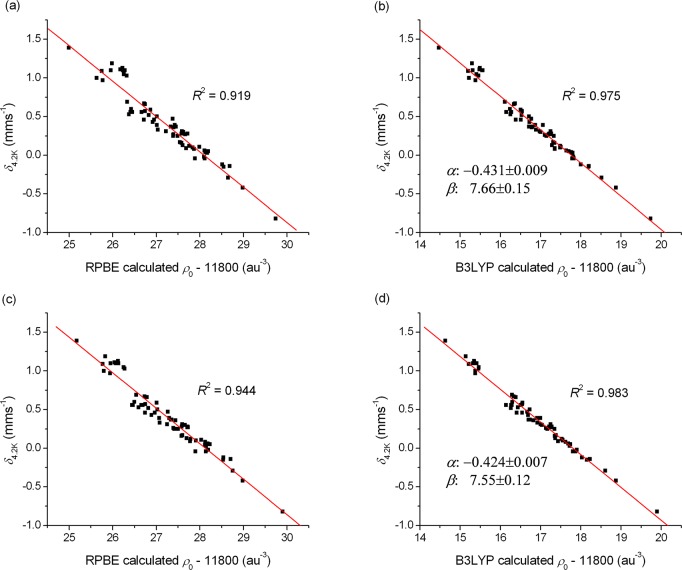
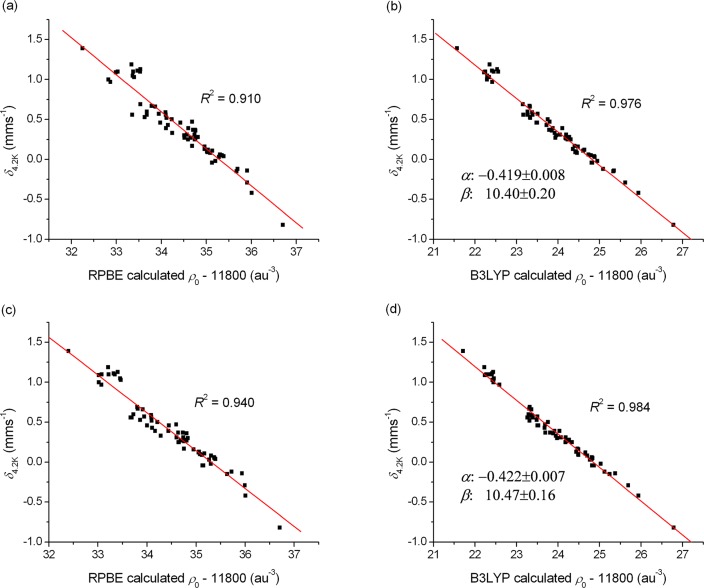
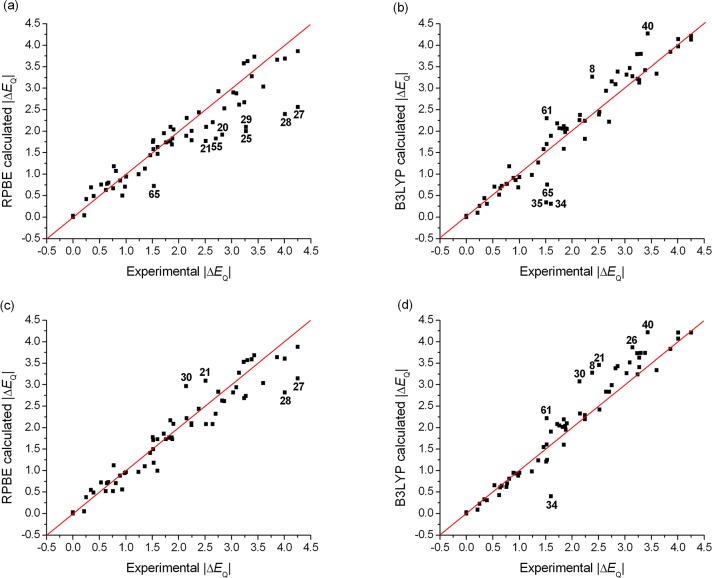
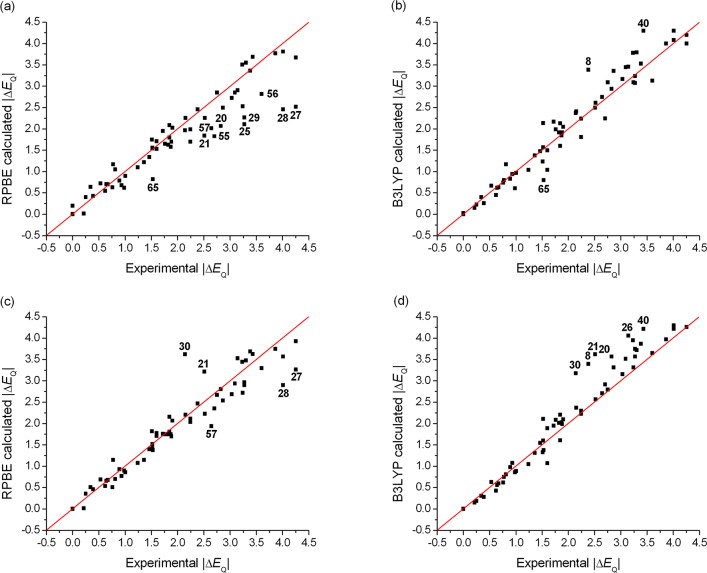
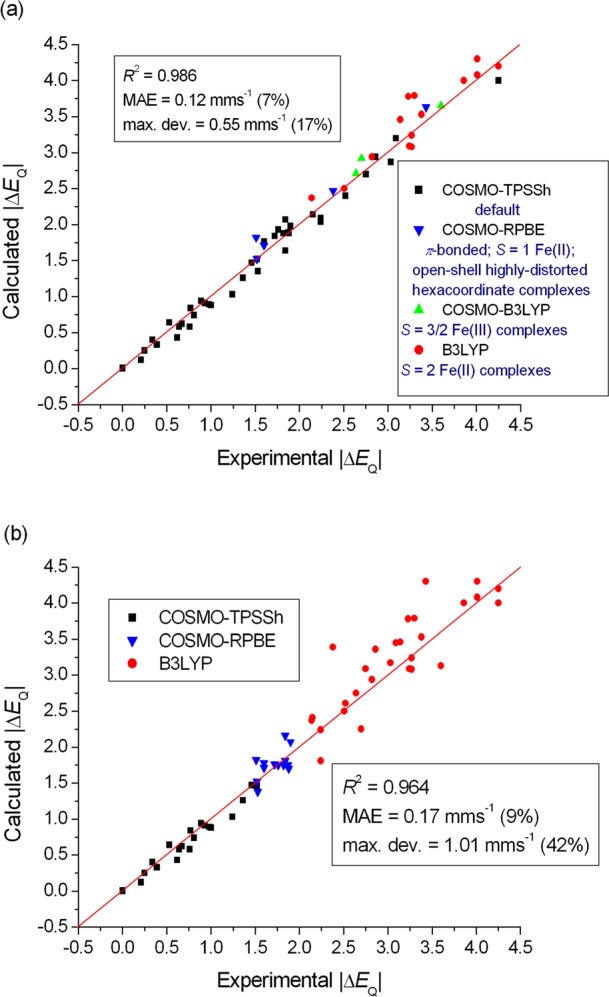
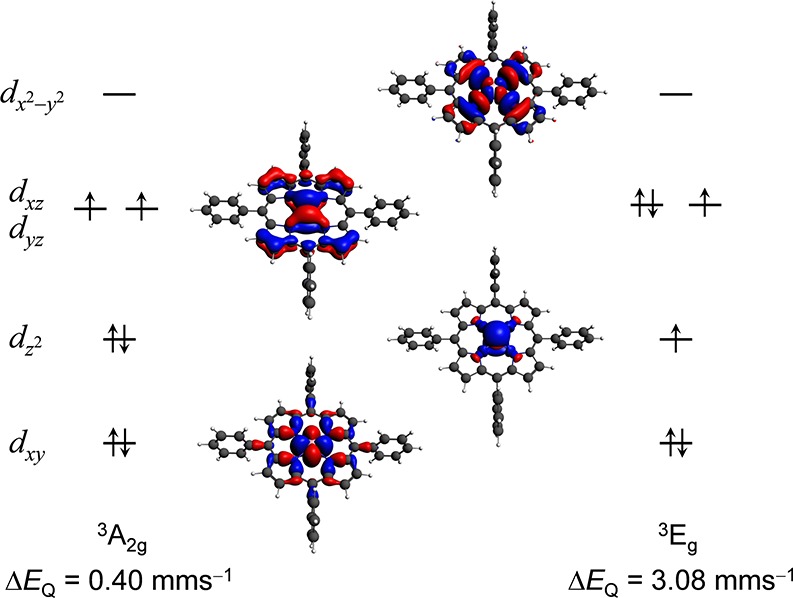
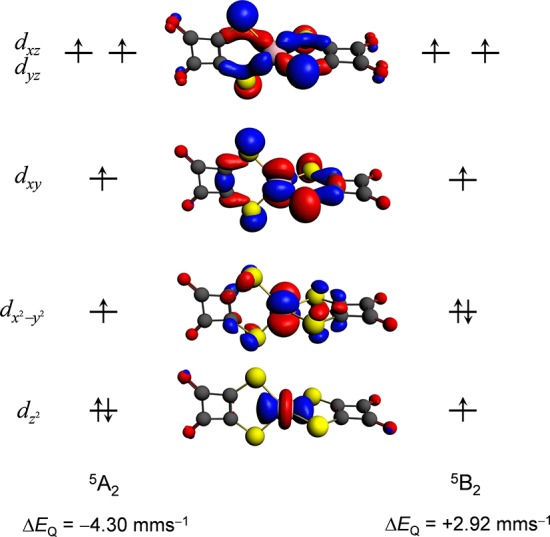
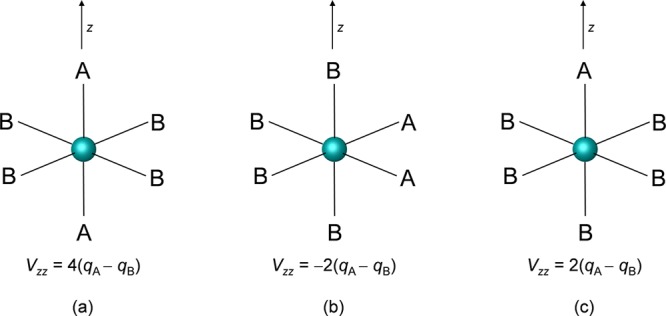
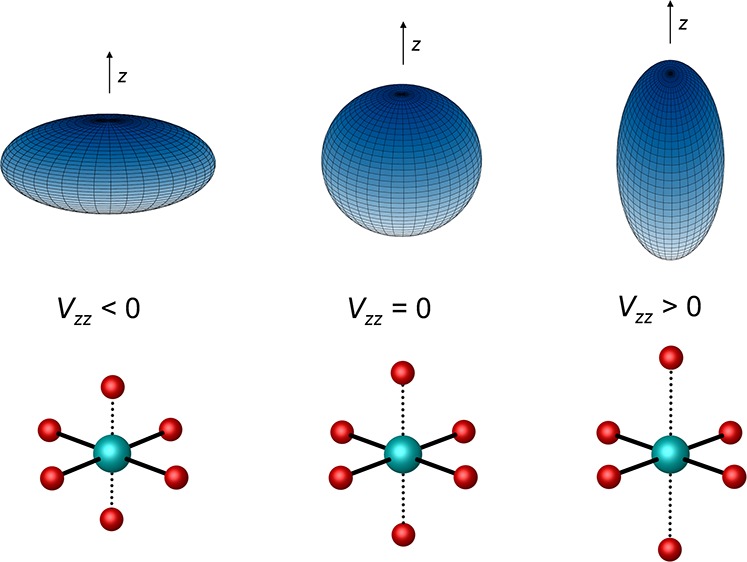
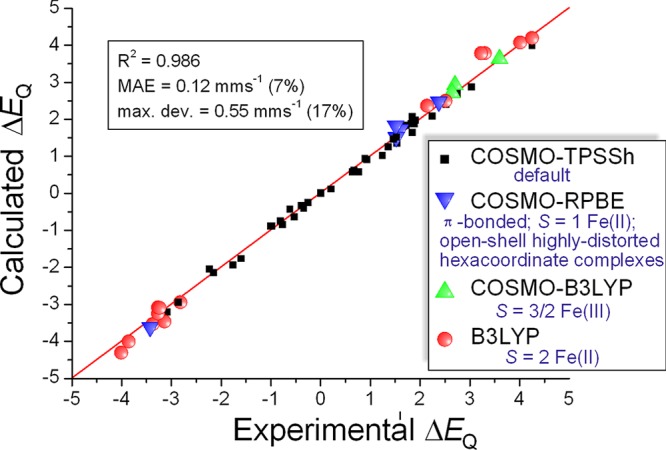
The performance of six frequently used density functional theory (DFT) methods (RPBE, OLYP, TPSS, B3LYP, B3LYP*, and TPSSh) in the prediction of Mössbauer isomer shifts(δ) and quadrupole splittings (ΔEQ) is studied for an extended and diverse set of Fe complexes. In addition to the influence of the applied density functional and the type of the basis set, the effect of the environment of the molecule, approximated with the conducting-like screening solvation model (COSMO) on the computed Mössbauer parameters, is also investigated. For the isomer shifts the COSMO-B3LYP method is found to provide accurate δ values for all 66 investigated complexes, with a mean absolute error (MAE) of 0.05 mm s-1 and a maximum deviation of 0.12 mm s-1. Obtaining accurate ΔEQ values presents a bigger challenge; however, with the selection of an appropriate DFT method, a reasonable agreement can be achieved between experiment and theory. Identifying the various chemical classes of compounds that need different treatment allowed us to construct a recipe for ΔEQ calculations; the application of this approach yields a MAE of 0.12 mm s-1 (7% error) and a maximum deviation of 0.55 mm s-1 (17% error). This accuracy should be sufficient for most chemical problems that concern Fe complexes. Furthermore, the reliability of the DFT approach is verified by extending the investigation to chemically relevant case studies which include geometric isomerism, phase transitions induced by variations of the electronic structure (e.g., spin crossover and inversion of the orbital ground state), and the description of electronically degenerate triplet and quintet states. Finally, the immense and often unexploited potential of utilizing the sign of the ΔEQ in characterizing distortions or in identifying the appropriate electronic state at the assignment of the spectral lines is also shown.
Figures
References
-
- Mössbauer R. L. Z. Phys. 1958, 151, 124–143.
-
- Greenwood N. N.; Gibb T. C.. Mössbauer Spectroscopy; Chapman and Hall: London, 1971.
-
- Vértes A.; Korecz L.; Burger K.. Mössbauer Spectroscopy; Elsevier Scientific Publishing Company: Amsterdam–Oxford–New York, 1979.
-
- Gütlich P.; Bill E.; Trautwein A. X.. Mössbauer Spectroscopy and Transition Metal Chemistry; Springer: Heidelberg–Dordrecht–London–New York, 2011.
-
- Hastings J. B.; Siddons D. P; van Bürck U.; Hollatz R.; Bergmann U. Phys. Rev. Lett. 1991, 66, 770–773. - PubMed
- Seto M.; Yoda Y.; Kikuta S.; Zhang X. W.; Ando M. Phys. Rev. Lett. 1995, 74, 3828–3831. - PubMed
- Sturhahn W.; Alp E. E.; Toellner T. S.; Hession P.; Hu M.; Sutter J. Hyperfine Interact. 1998, 113, 47–58.
- Rüffer R.; Chumakov A. I. Hyperfine Interact. 2000, 128, 255–272.
- Sergueev I.; van Bürck U.; Chumakov A. I.; Asthalter T.; Smirnov G. V.; Franz H.; Rüffer R.; Petry W. Phys. Rev. B 2006, 73, 24203.
- Potapkin V.; Chumakov A. I.; Smirnov G. V.; Celse J. P.; Rüffer R.; McCammon C.; Dubrovinsky L. J. Synchrotron Radiat. 2012, 19, 559–569. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources