

Rab6 regulation of the kinesin family KIF1C motor domain contributes to Golgi tethering
- PMID: 25821985
- PMCID: PMC4405695
- DOI: 10.7554/eLife.06029
Rab6 regulation of the kinesin family KIF1C motor domain contributes to Golgi tethering
Abstract
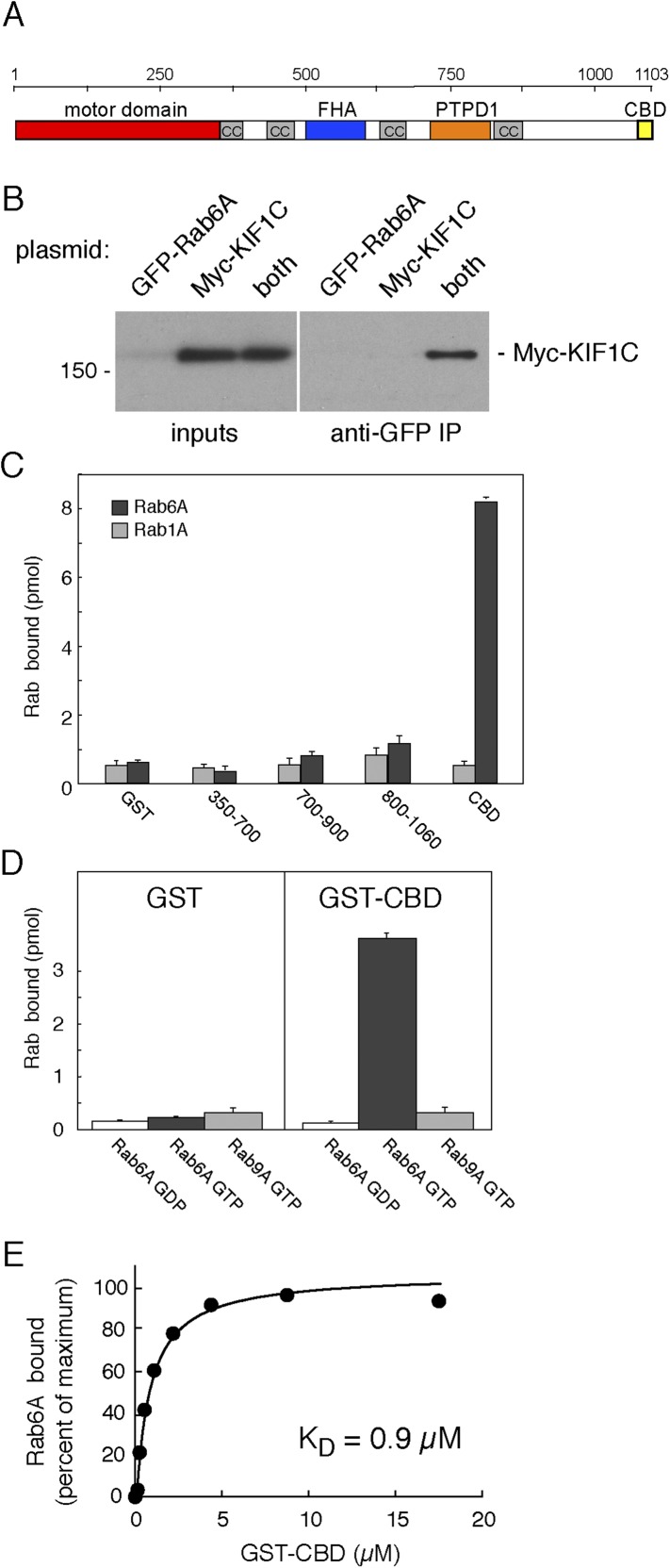
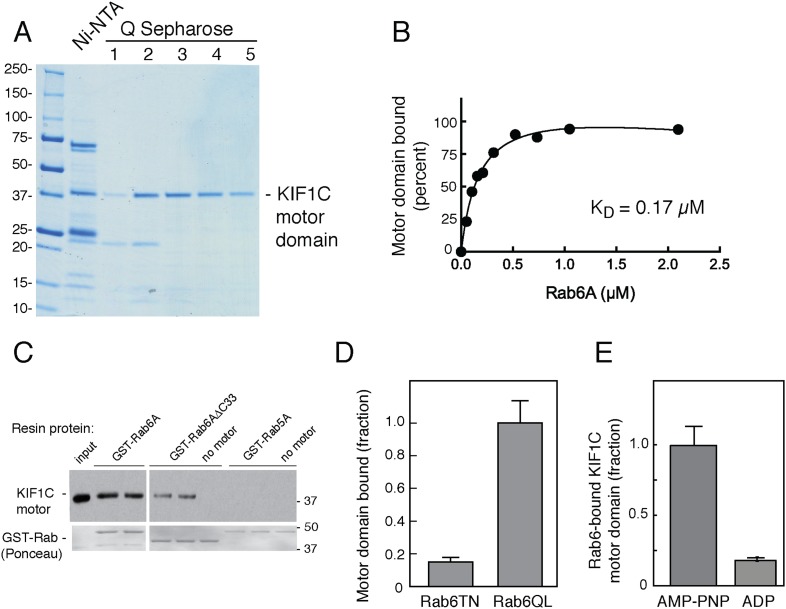
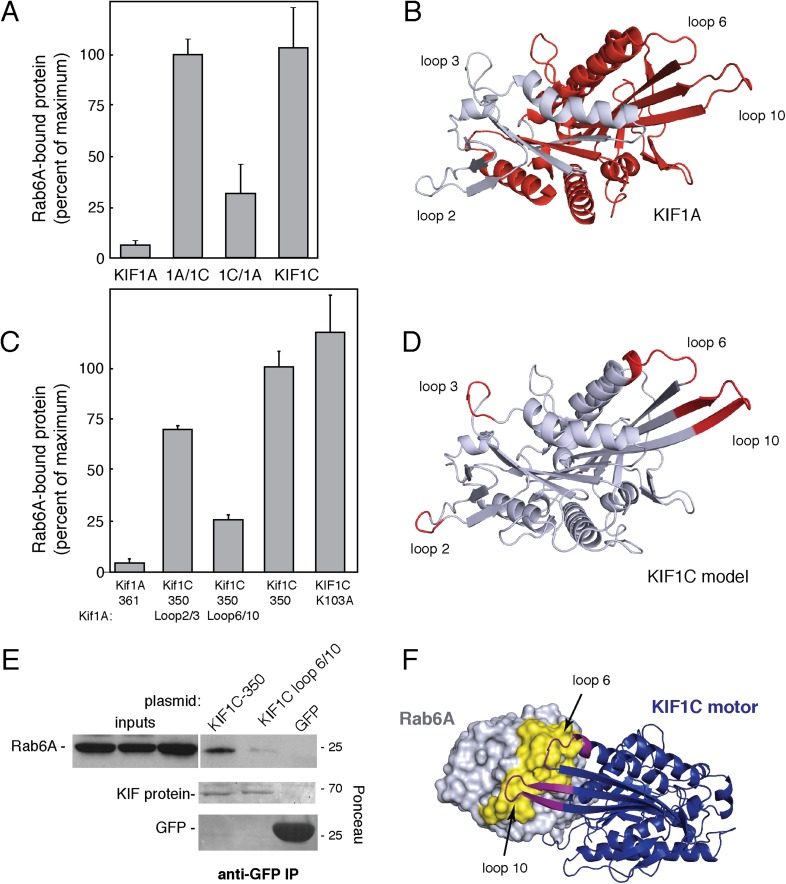
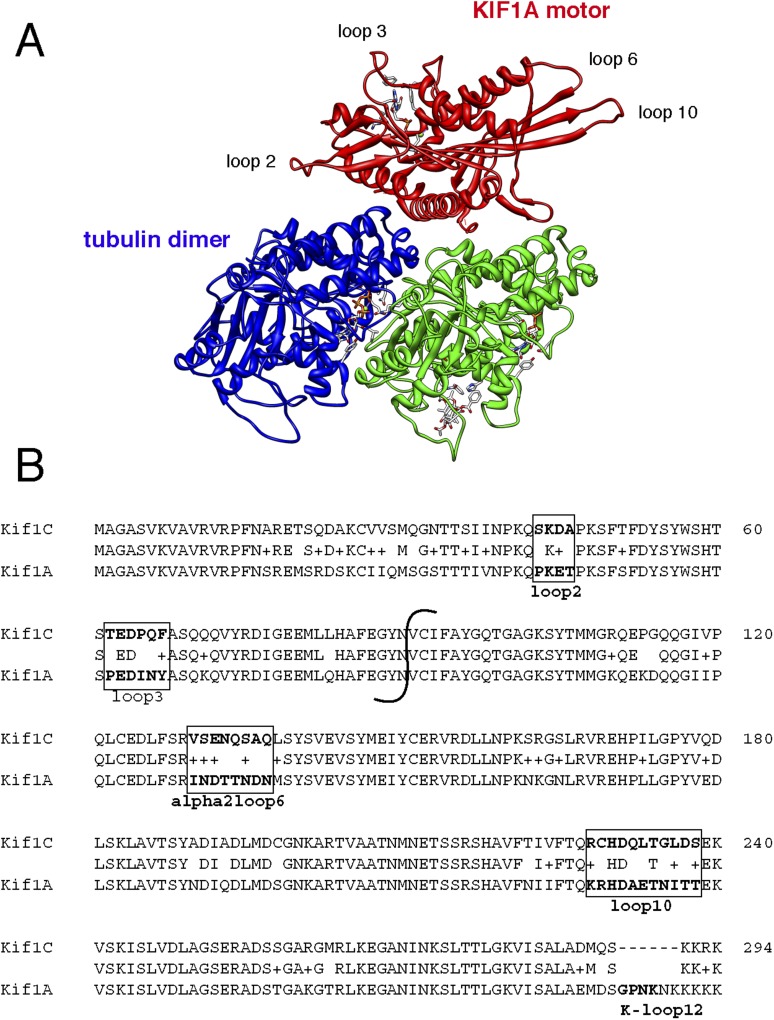
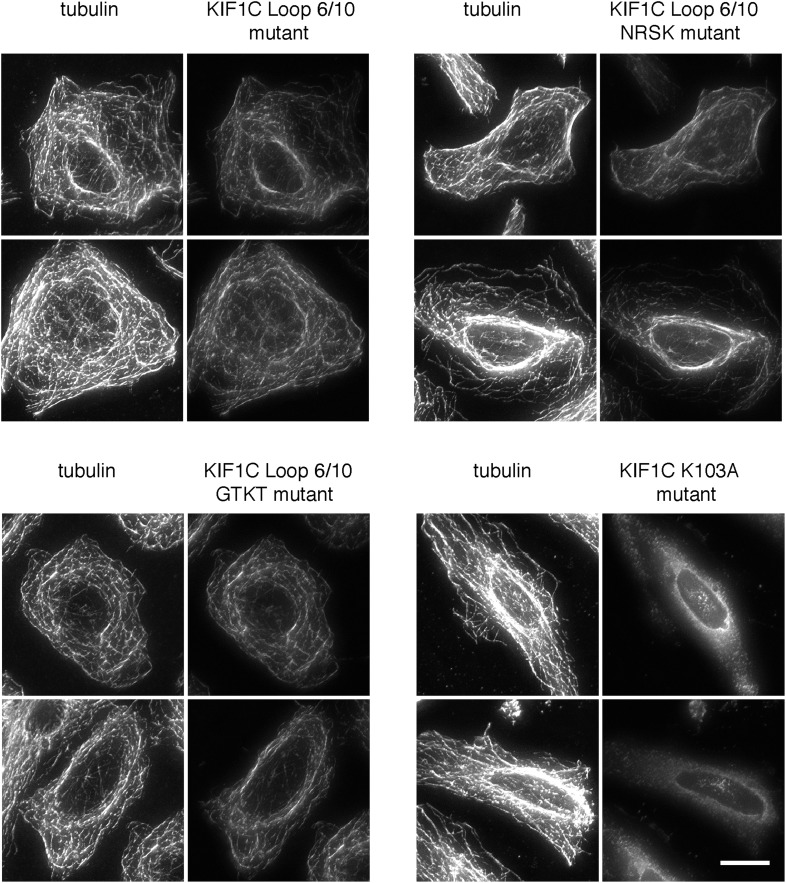
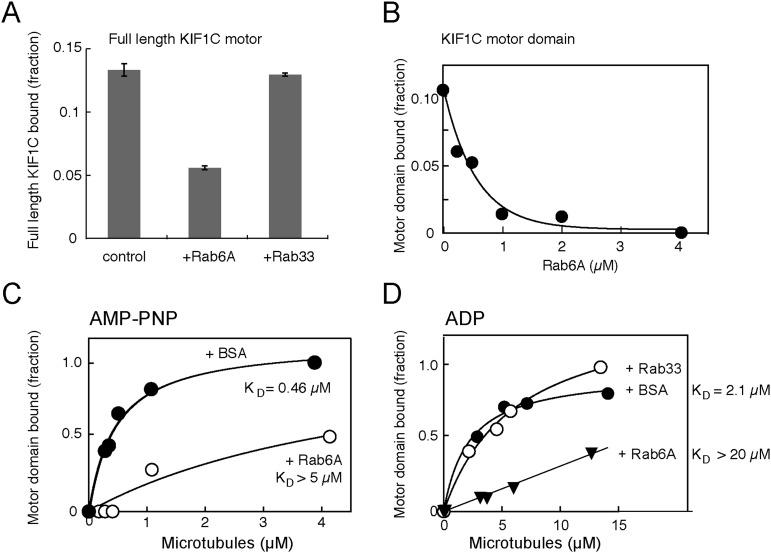
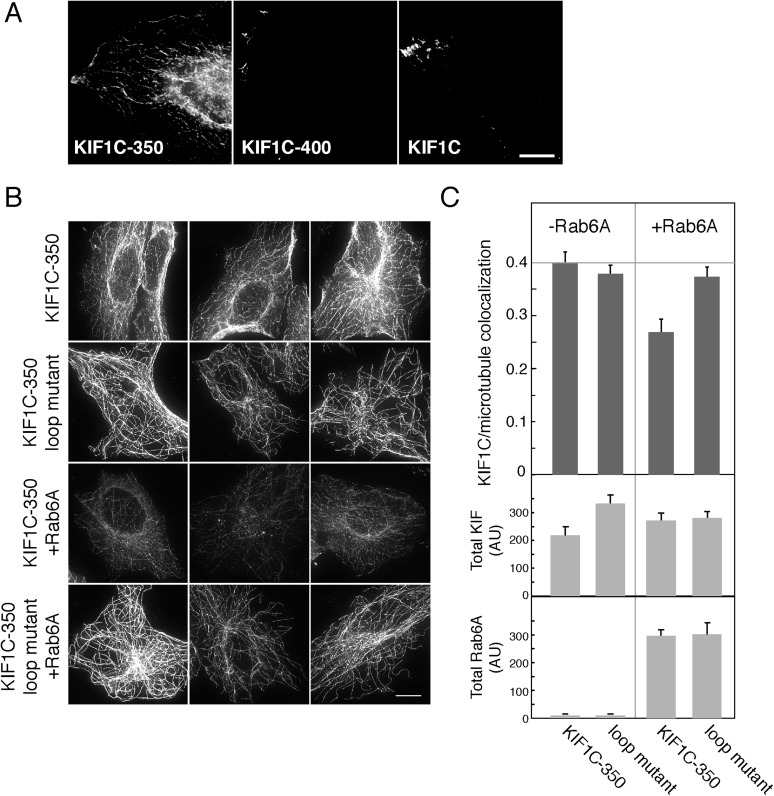
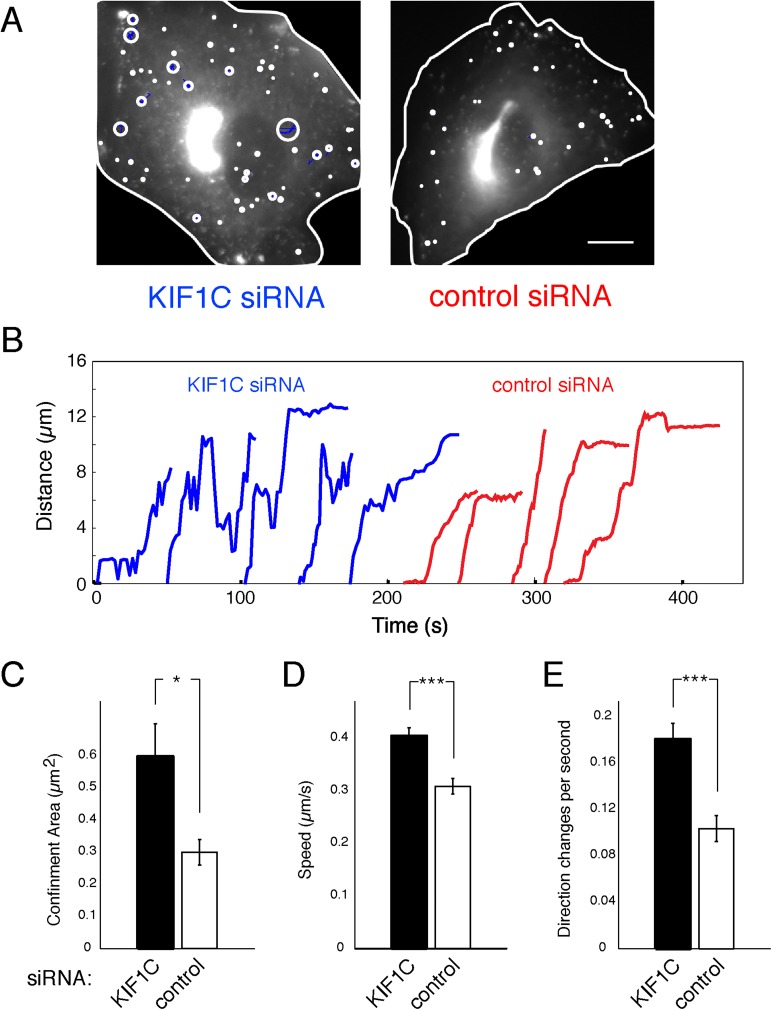
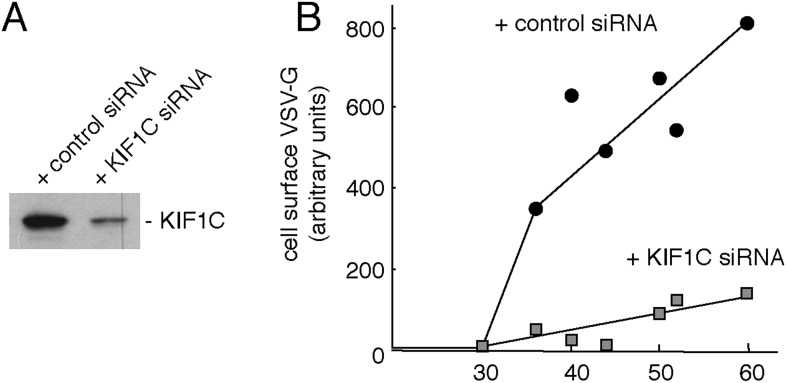
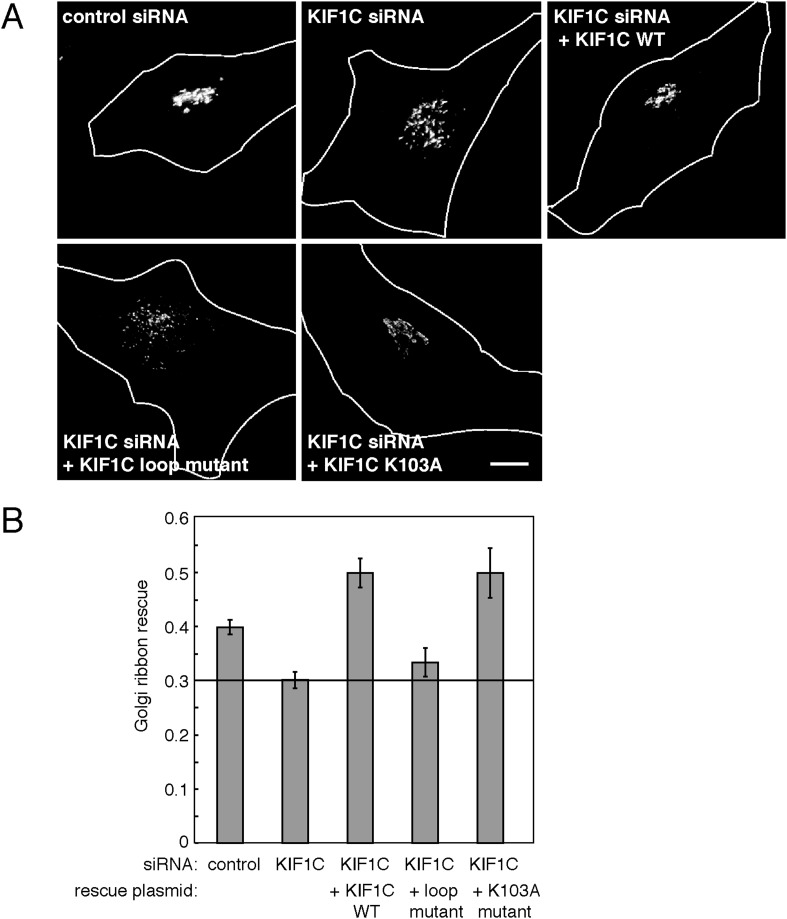
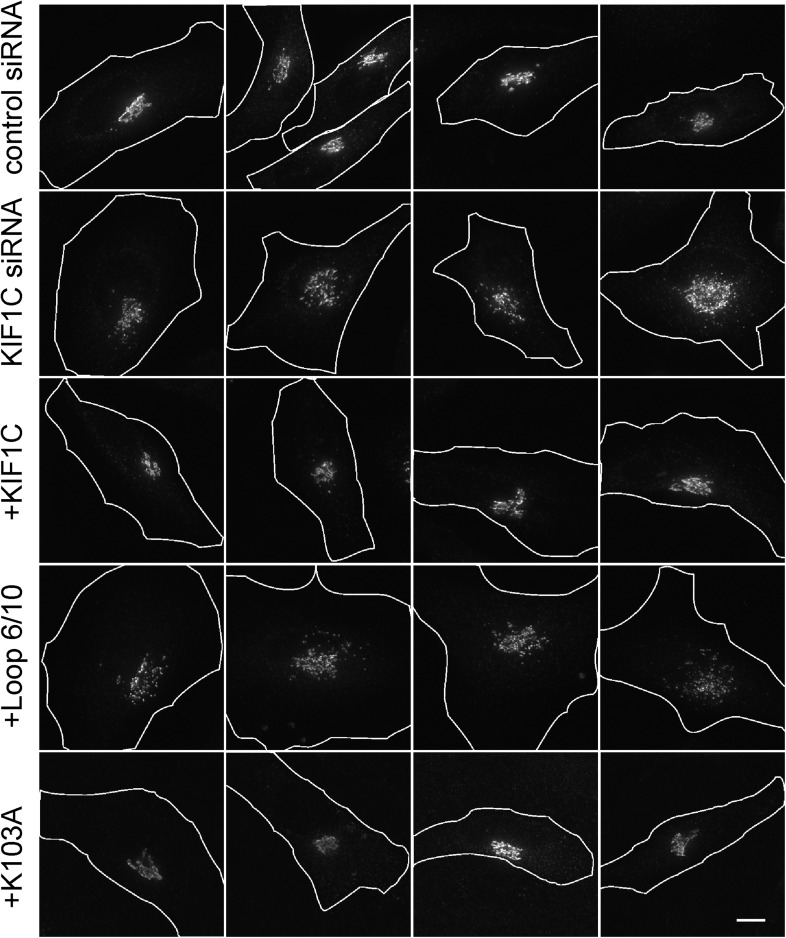
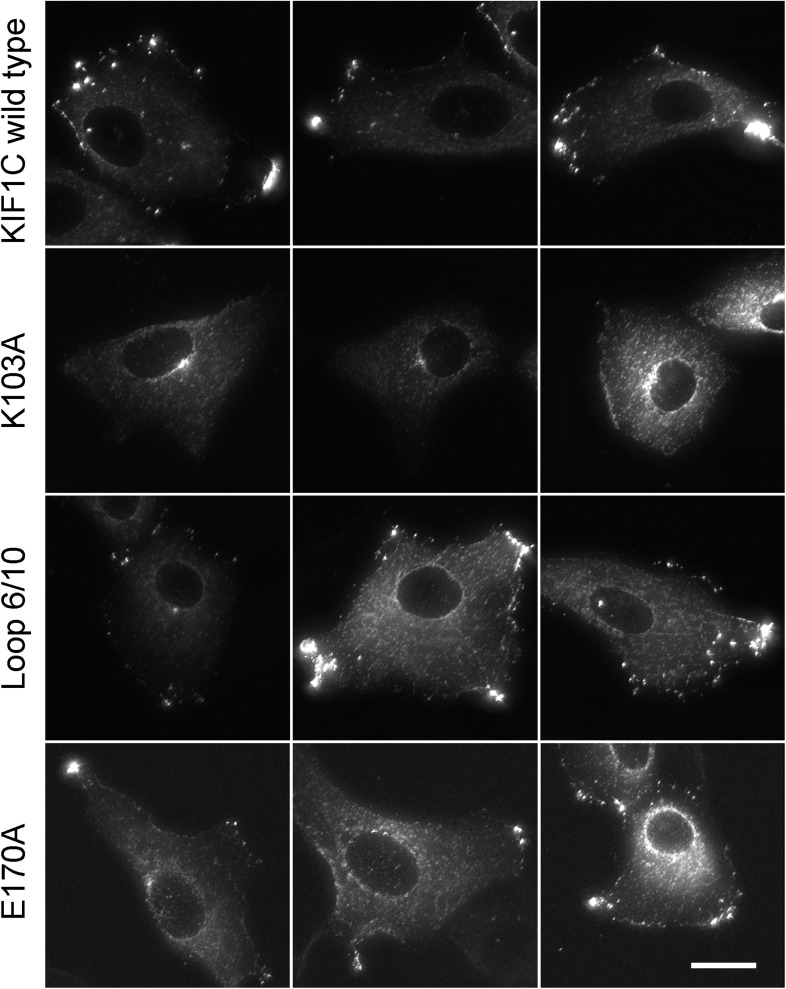
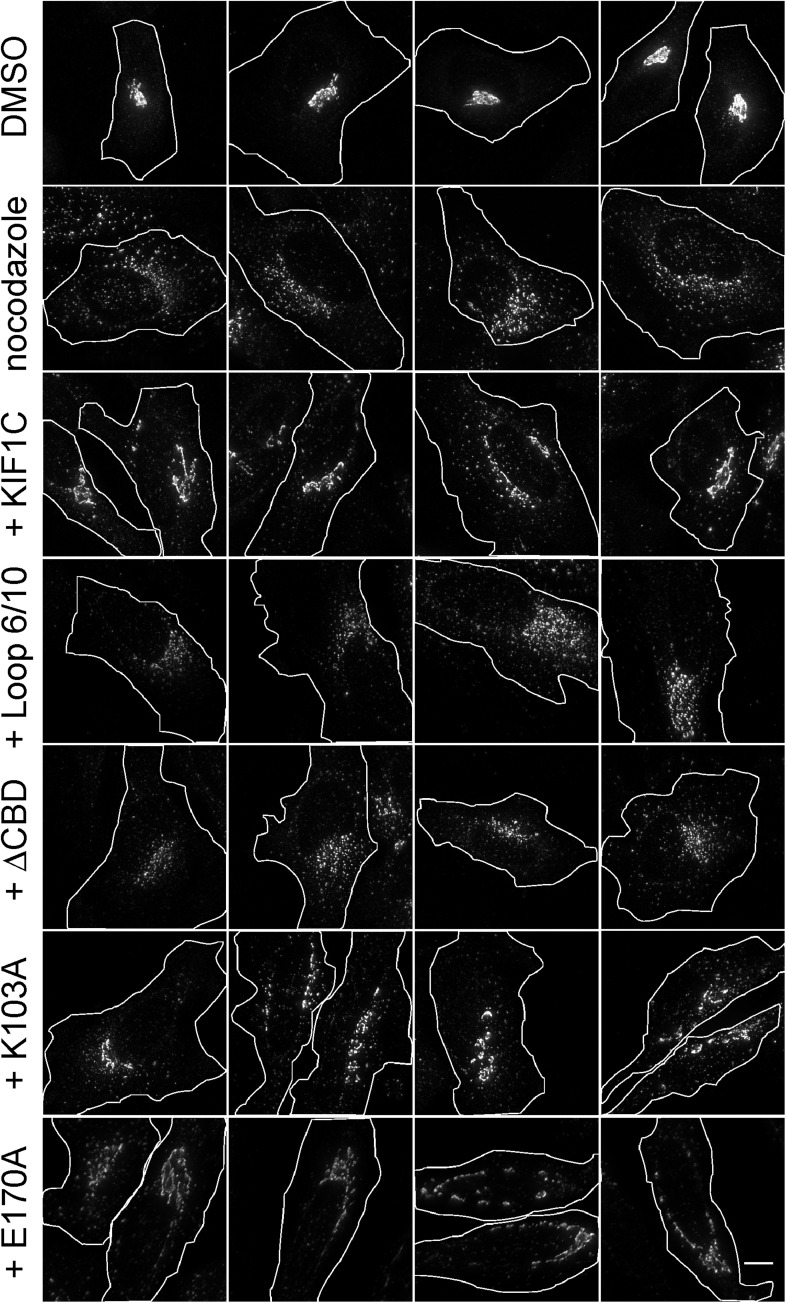
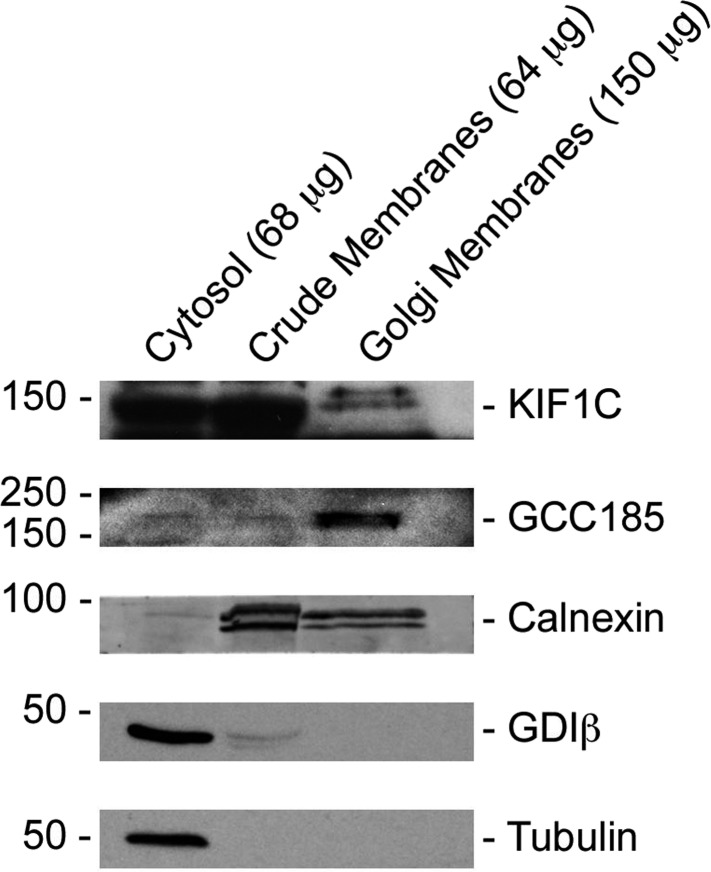
Most kinesins transport cargoes bound to their C-termini and use N-terminal motor domains to move along microtubules. We report here a novel function for KIF1C: it transports Rab6A-vesicles and can influence Golgi complex organization. These activities correlate with KIF1C's capacity to bind the Golgi protein Rab6A directly, both via its motor domain and C-terminus. Rab6A binding to the motor domain inhibits microtubule interaction in vitro and in cells, decreasing the amount of motile KIF1C. KIF1C depletion slows protein delivery to the cell surface, interferes with vesicle motility, and triggers Golgi fragmentation. KIF1C can protect Golgi membranes from fragmentation in cells lacking an intact microtubule network. Rescue of fragmentation requires sequences that enable KIF1C to bind Rab6A at both ends, but not KIF1C motor function. Rab6A binding to KIF1C's motor domain represents an entirely new mode of regulation for a kinesin motor, and likely has important consequences for KIF1C's cellular functions.
Keywords: Golgi complex; KIF1C; Rab GTPase; biochemistry; cell biology; kinesin; microtubule motor; none.
Conflict of interest statement
SRP: Reviewing editor,
The other authors declare that no competing interests exist.
Figures



References
-
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. - DOI - PMC - PubMed
-
- del Rio R, McAllister RD, Meeker ND, Wall EH, Bond JP, Kyttaris VC, Tsokos GC, Tung KS, Teuscher C. Identification of Orch3, a locus controlling dominant resistance to autoimmune orchitis, as kinesin family member 1C. PLOS Genetics. 2012;8:e1003140. doi: 10.1371/journal.pgen.1003140. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
