

Parkin cooperates with GDNF/RET signaling to prevent dopaminergic neuron degeneration
- PMID: 25822020
- PMCID: PMC4611569
- DOI: 10.1172/JCI79300
Parkin cooperates with GDNF/RET signaling to prevent dopaminergic neuron degeneration
Abstract
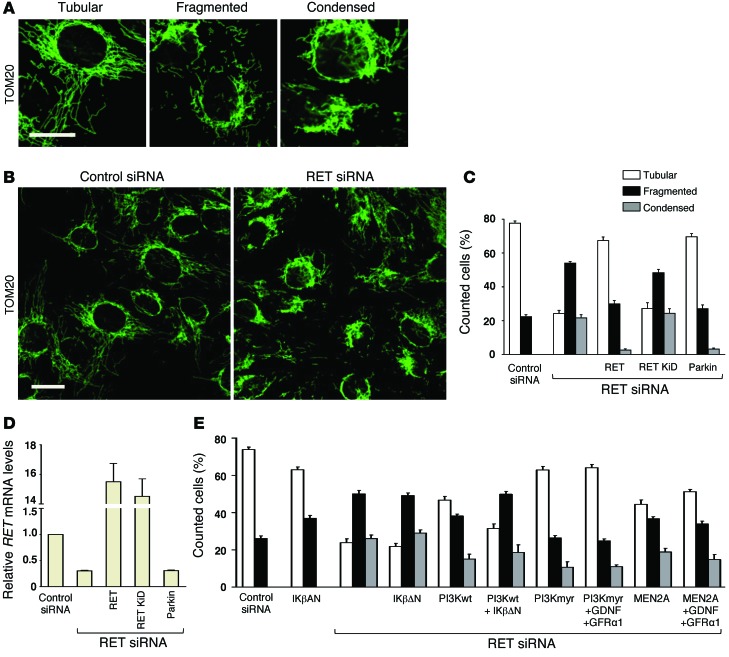
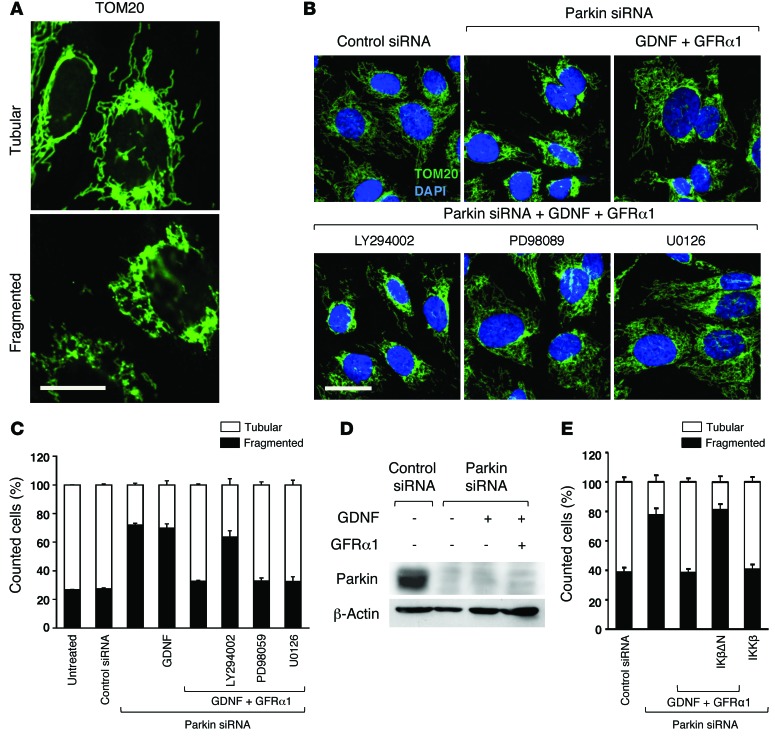
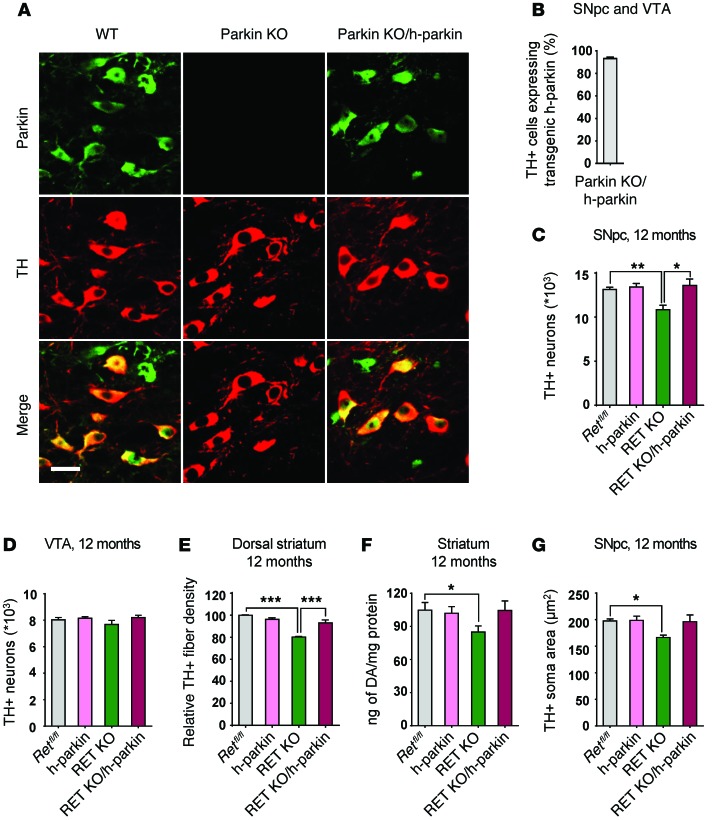
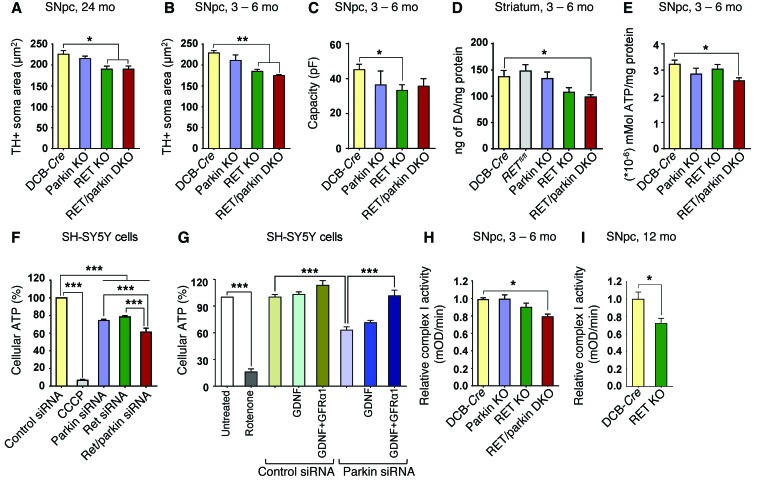
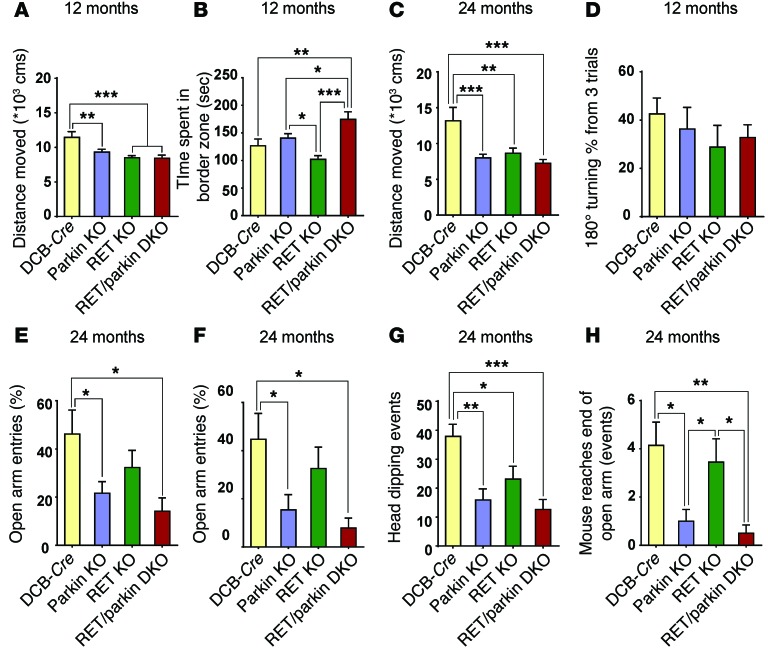
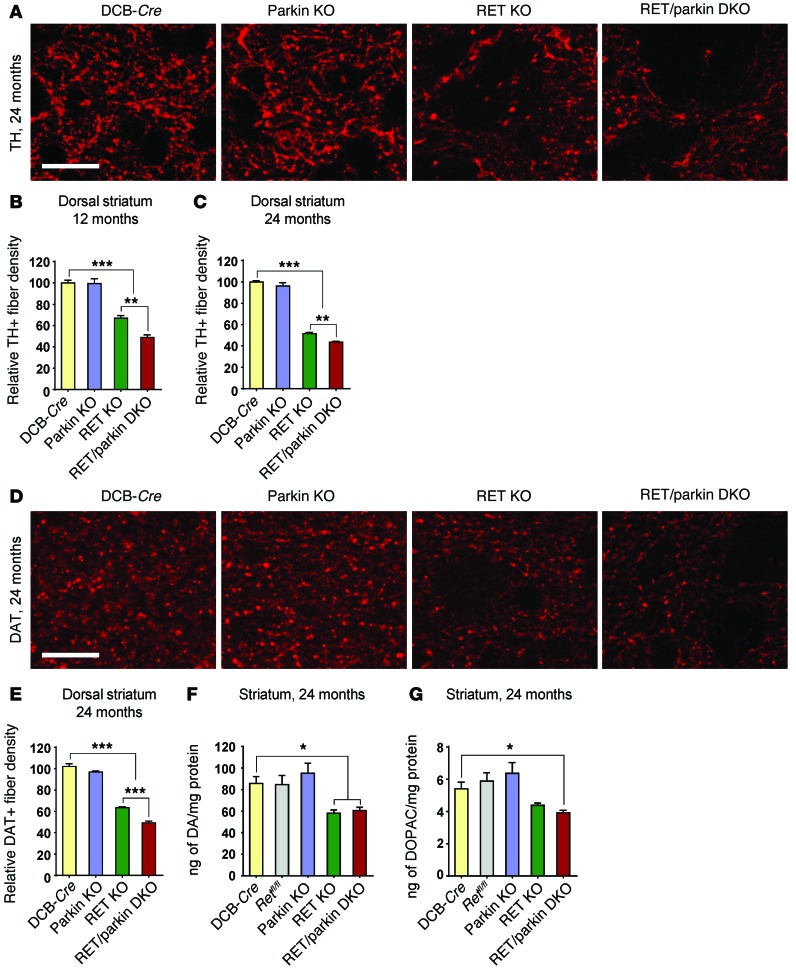
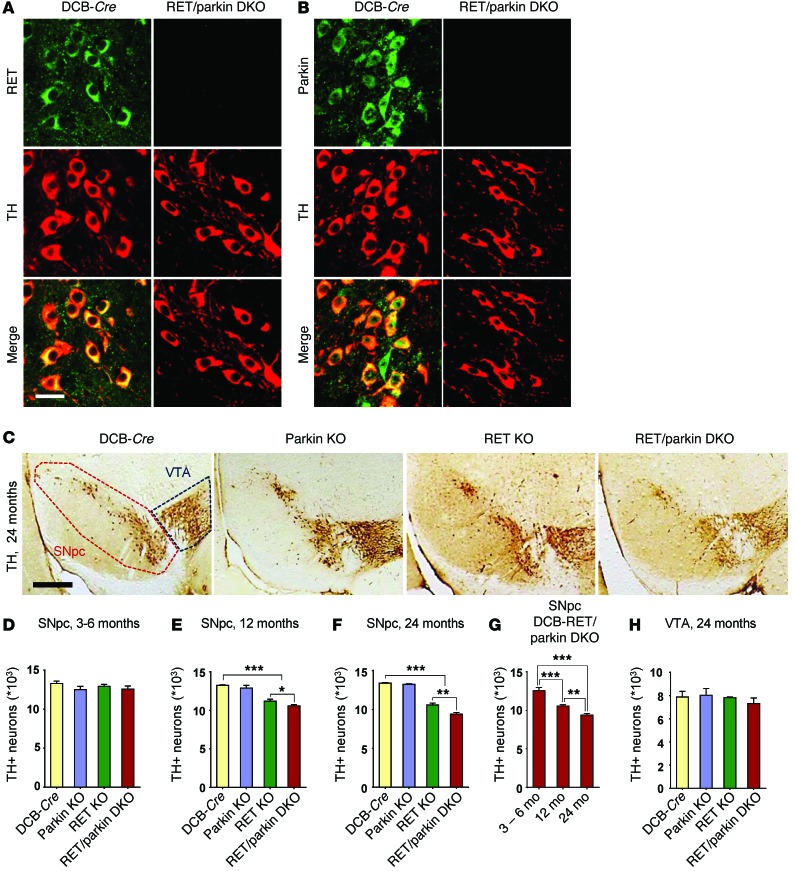
Parkin and the glial cell line-derived neurotrophic factor (GDNF) receptor RET have both been independently linked to the dopaminergic neuron degeneration that underlies Parkinson's disease (PD). In the present study, we demonstrate that there is genetic crosstalk between parkin and the receptor tyrosine kinase RET in two different mouse models of PD. Mice lacking both parkin and RET exhibited accelerated dopaminergic cell and axonal loss compared with parkin-deficient animals, which showed none, and RET-deficient mice, in which we found moderate degeneration. Transgenic expression of parkin protected the dopaminergic systems of aged RET-deficient mice. Downregulation of either parkin or RET in neuronal cells impaired mitochondrial function and morphology. Parkin expression restored mitochondrial function in GDNF/RET-deficient cells, while GDNF stimulation rescued mitochondrial defects in parkin-deficient cells. In both cases, improved mitochondrial function was the result of activation of the prosurvival NF-κB pathway, which was mediated by RET through the phosphoinositide-3-kinase (PI3K) pathway. Taken together, these observations indicate that parkin and the RET signaling cascade converge to control mitochondrial integrity and thereby properly maintain substantia nigra pars compacta dopaminergic neurons and their innervation in the striatum. The demonstration of crosstalk between parkin and RET highlights the interplay in the protein network that is altered in PD and suggests potential therapeutic targets and strategies to treat PD.
Figures
Similar articles
-
Ret is essential to mediate GDNF's neuroprotective and neuroregenerative effect in a Parkinson disease mouse model.Cell Death Dis. 2016 Sep 8;7(9):e2359. doi: 10.1038/cddis.2016.263. Cell Death Dis. 2016. PMID: 27607574 Free PMC article.
-
Electroacupuncture Therapy Ameliorates Motor Dysfunction via Brain-Derived Neurotrophic Factor and Glial Cell Line-Derived Neurotrophic Factor in a Mouse Model of Parkinson's Disease.J Gerontol A Biol Sci Med Sci. 2020 Mar 9;75(4):712-721. doi: 10.1093/gerona/glz256. J Gerontol A Biol Sci Med Sci. 2020. PMID: 31644786
-
Lack of Parkin Anticipates the Phenotype and Affects Mitochondrial Morphology and mtDNA Levels in a Mouse Model of Parkinson's Disease.J Neurosci. 2018 Jan 24;38(4):1042-1053. doi: 10.1523/JNEUROSCI.1384-17.2017. Epub 2017 Dec 8. J Neurosci. 2018. PMID: 29222404 Free PMC article.
-
GDNF-Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease.FEBS Lett. 2015 Dec 21;589(24 Pt A):3760-72. doi: 10.1016/j.febslet.2015.11.006. Epub 2015 Nov 7. FEBS Lett. 2015. PMID: 26555190 Review.
-
GDNF/RET signaling in dopamine neurons in vivo.Cell Tissue Res. 2020 Oct;382(1):135-146. doi: 10.1007/s00441-020-03268-9. Epub 2020 Sep 1. Cell Tissue Res. 2020. PMID: 32870383 Review.
Cited by
-
Is activation of GDNF/RET signaling the answer for successful treatment of Parkinson's disease? A discussion of data from the culture dish to the clinic.Neural Regen Res. 2022 Jul;17(7):1462-1467. doi: 10.4103/1673-5374.327330. Neural Regen Res. 2022. PMID: 34916419 Free PMC article. Review.
-
The neuroprotective and regenerative potential of parkin and GDNF/Ret signaling in the midbrain dopaminergic system.Neural Regen Res. 2015 Nov;10(11):1752-3. doi: 10.4103/1673-5374.165295. Neural Regen Res. 2015. PMID: 26807104 Free PMC article. No abstract available.
-
Reduced expression of PARK2 in manganese-exposed smelting workers.Neurotoxicology. 2017 Sep;62:258-264. doi: 10.1016/j.neuro.2017.08.006. Epub 2017 Aug 18. Neurotoxicology. 2017. PMID: 28826884 Free PMC article.
-
Ret is essential to mediate GDNF's neuroprotective and neuroregenerative effect in a Parkinson disease mouse model.Cell Death Dis. 2016 Sep 8;7(9):e2359. doi: 10.1038/cddis.2016.263. Cell Death Dis. 2016. PMID: 27607574 Free PMC article.
-
Neuroprotective Transcription Factors in Animal Models of Parkinson Disease.Neural Plast. 2016;2016:6097107. doi: 10.1155/2016/6097107. Epub 2015 Dec 31. Neural Plast. 2016. PMID: 26881122 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
