

MEL-18 loss mediates estrogen receptor-α downregulation and hormone independence
- PMID: 25822021
- PMCID: PMC4463188
- DOI: 10.1172/JCI73743
MEL-18 loss mediates estrogen receptor-α downregulation and hormone independence
Abstract
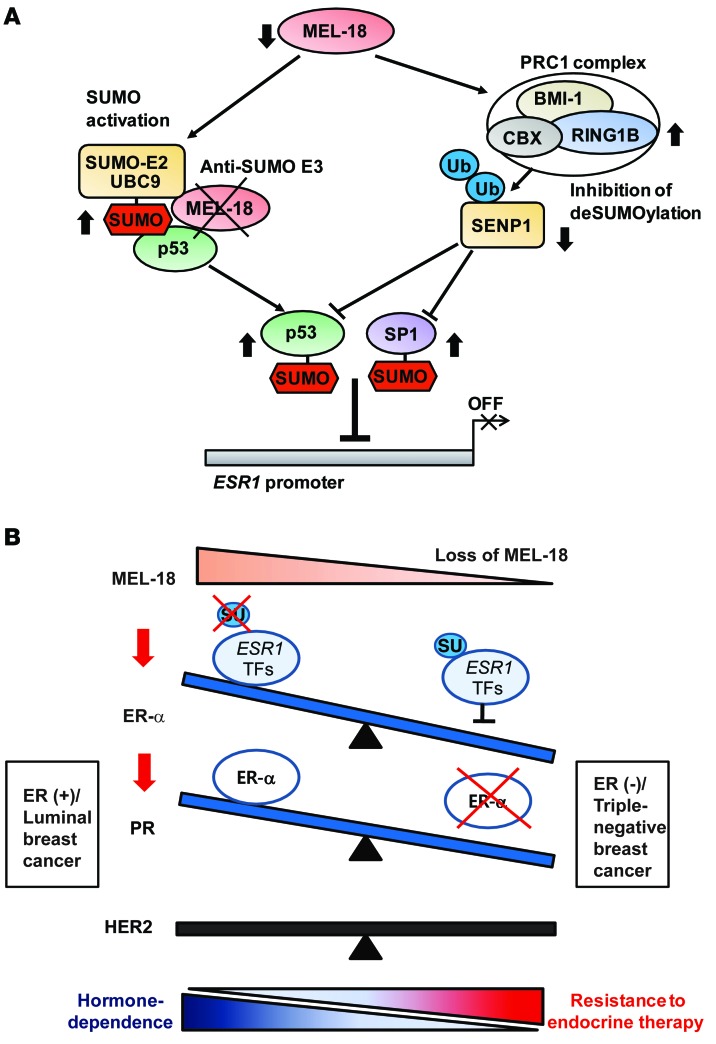
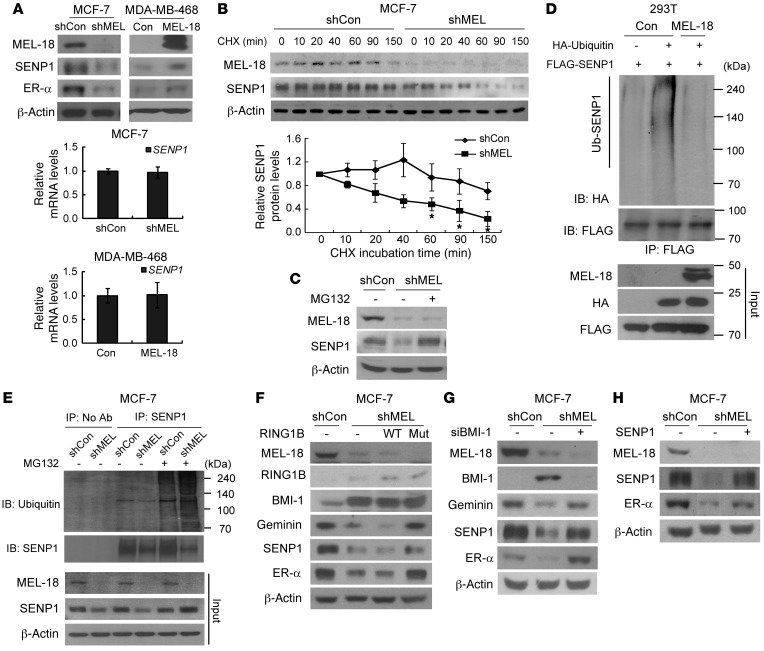
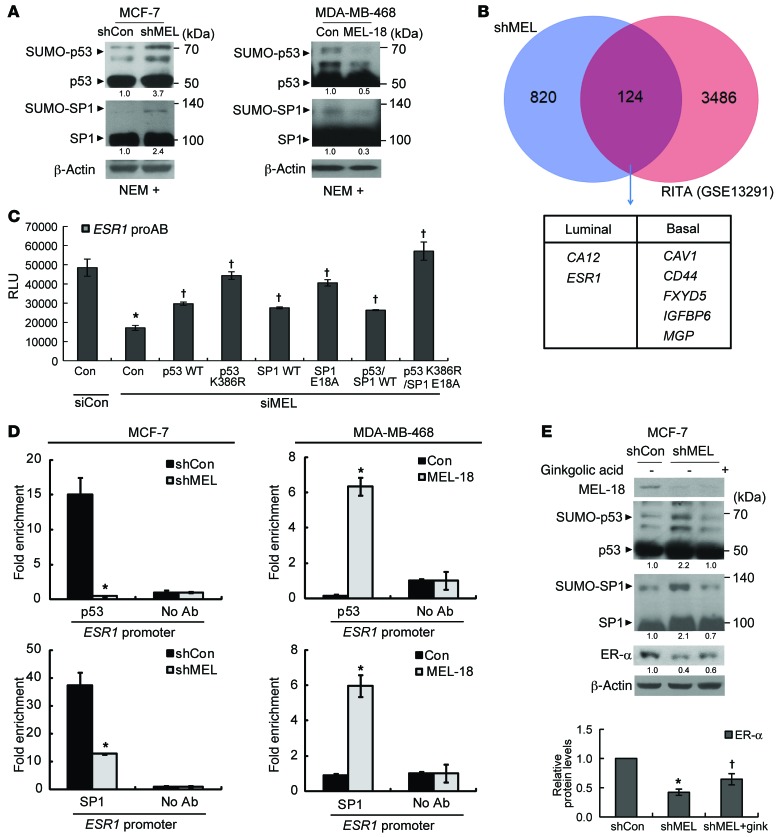
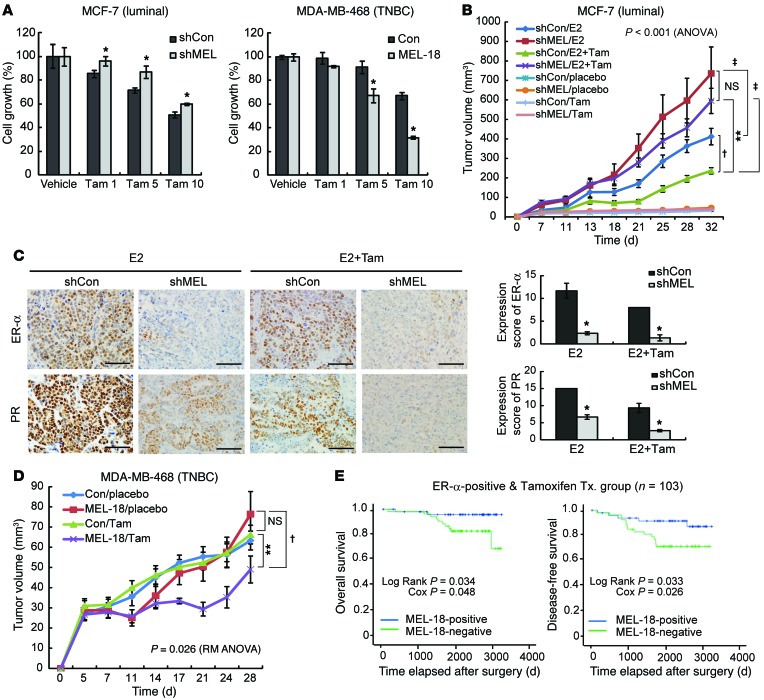
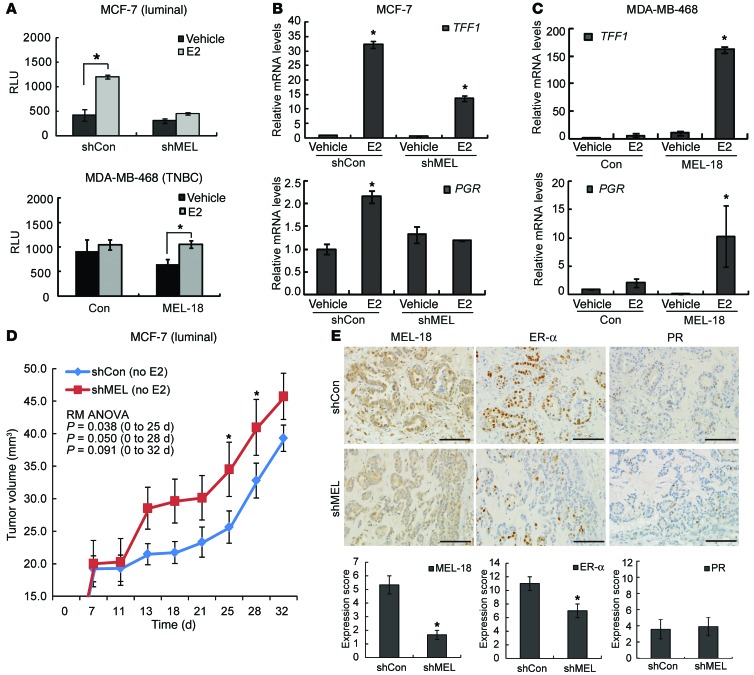
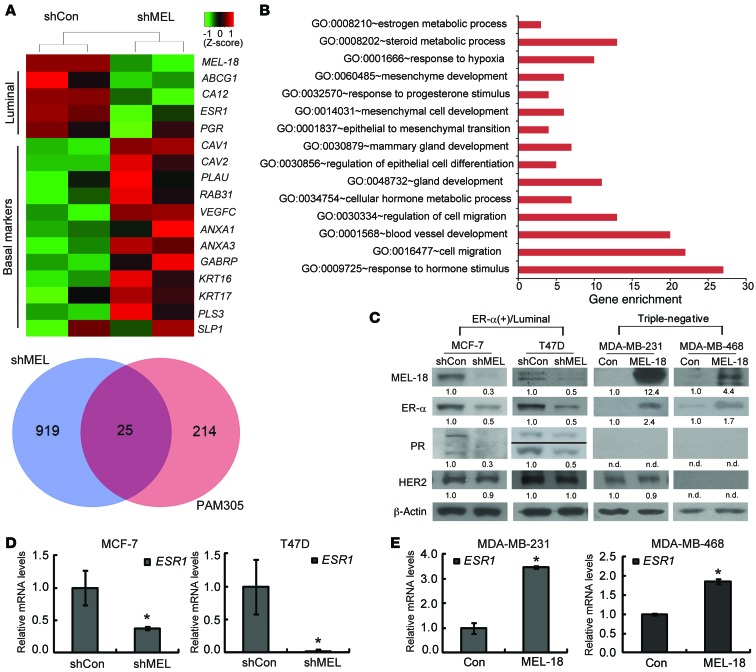
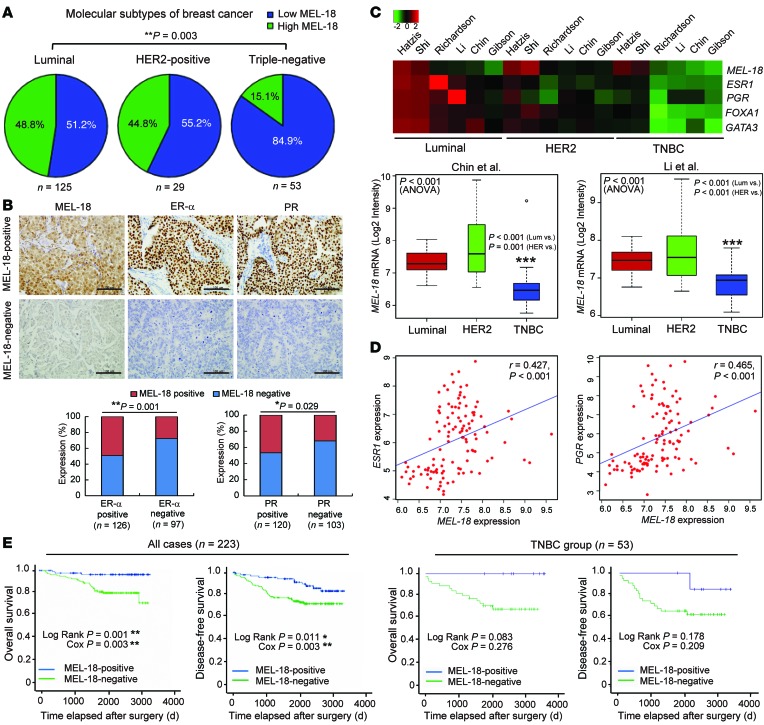
The polycomb protein MEL-18 has been proposed as a tumor suppressor in breast cancer; however, its functional relevance to the hormonal regulation of breast cancer remains unknown. Here, we demonstrated that MEL-18 loss contributes to the hormone-independent phenotype of breast cancer by modulating hormone receptor expression. In multiple breast cancer cohorts, MEL-18 was markedly downregulated in triple-negative breast cancer (TNBC). MEL-18 expression positively correlated with the expression of luminal markers, including estrogen receptor-α (ER-α, encoded by ESR1). MEL-18 loss was also associated with poor response to antihormonal therapy in ER-α-positive breast cancer. Furthermore, whereas MEL-18 loss in luminal breast cancer cells resulted in the downregulation of expression and activity of ER-α and the progesterone receptor (PR), MEL-18 overexpression restored ER-α expression in TNBC. Consistently, in vivo xenograft experiments demonstrated that MEL-18 loss induces estrogen-independent growth and tamoxifen resistance in luminal breast cancer, and that MEL-18 overexpression confers tamoxifen sensitivity in TNBC. MEL-18 suppressed SUMOylation of the ESR1 transactivators p53 and SP1, thereby driving ESR1 transcription. MEL-18 facilitated the deSUMOylation process by inhibiting BMI-1/RING1B-mediated ubiquitin-proteasomal degradation of SUMO1/sentrin-specific protease 1 (SENP1). These findings demonstrate that MEL-18 is a SUMO-dependent regulator of hormone receptors and suggest MEL-18 expression as a marker for determining the antihormonal therapy response in patients with breast cancer.
Figures
Similar articles
-
IGF-II mRNA expression in breast cancer: predictive value and relationship to other prognostic factors.Int J Biol Markers. 2010 Jul-Sep;25(3):150-6. doi: 10.1177/172460081002500305. Int J Biol Markers. 2010. PMID: 20677165
-
Harmful effect of ERβ on BCRP-mediated drug resistance and cell proliferation in ERα/PR-negative breast cancer.FEBS J. 2013 Dec;280(23):6128-40. doi: 10.1111/febs.12533. Epub 2013 Oct 8. FEBS J. 2013. PMID: 24103091
-
A candidate molecular signature associated with tamoxifen failure in primary breast cancer.Breast Cancer Res. 2008;10(5):R88. doi: 10.1186/bcr2158. Epub 2008 Oct 17. Breast Cancer Res. 2008. PMID: 18928543 Free PMC article.
-
Clinical significance of estrogen receptor phosphorylation.Endocr Relat Cancer. 2011 Jan 19;18(1):R1-14. doi: 10.1677/ERC-10-0070. Print 2011 Feb. Endocr Relat Cancer. 2011. PMID: 21149515 Review.
-
[Progesterone receptor in breast cancer: fundamental data and clinical applications].Bull Cancer. 1996;83 Suppl 1:1-5. Bull Cancer. 1996. PMID: 8777338 Review. French. No abstract available.
Cited by
-
The role of C1orf50 in breast cancer progression and prognosis.Breast Cancer. 2025 Mar;32(2):292-305. doi: 10.1007/s12282-024-01653-8. Epub 2024 Nov 28. Breast Cancer. 2025. PMID: 39604563 Free PMC article.
-
SUMO proteins in the cardiovascular system: friend or foe?J Biomed Sci. 2020 Oct 24;27(1):98. doi: 10.1186/s12929-020-00689-0. J Biomed Sci. 2020. PMID: 33099299 Free PMC article. Review.
-
MEL-18, a tumor suppressor for aggressive breast cancer.Oncotarget. 2015 Jun 30;6(18):15710-1. doi: 10.18632/oncotarget.4565. Oncotarget. 2015. PMID: 26158291 Free PMC article. No abstract available.
-
Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms.Nat Commun. 2018 Aug 23;9(1):3377. doi: 10.1038/s41467-018-05728-x. Nat Commun. 2018. PMID: 30139998 Free PMC article.
-
Bone metastasis pattern in initial metastatic breast cancer: a population-based study.Cancer Manag Res. 2018 Feb 9;10:287-295. doi: 10.2147/CMAR.S155524. eCollection 2018. Cancer Manag Res. 2018. PMID: 29467583 Free PMC article.
References
-
- Keen JC, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat. 2003;81(3):177–186. doi: 10.1023/A:1026146524737. - DOI - PubMed
-
- Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 2001;61(19):7025–7029. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
