

Massively Parallel Functional Analysis of BRCA1 RING Domain Variants
- PMID: 25823446
- PMCID: PMC4492368
- DOI: 10.1534/genetics.115.175802
Massively Parallel Functional Analysis of BRCA1 RING Domain Variants
Erratum in
-
Massively Parallel Functional Analysis of BRCA1 RING Domain Variants.Genetics. 2017 Dec;207(4):1713. doi: 10.1534/genetics.117.300355. Genetics. 2017. PMID: 29203703 Free PMC article. No abstract available.
Abstract
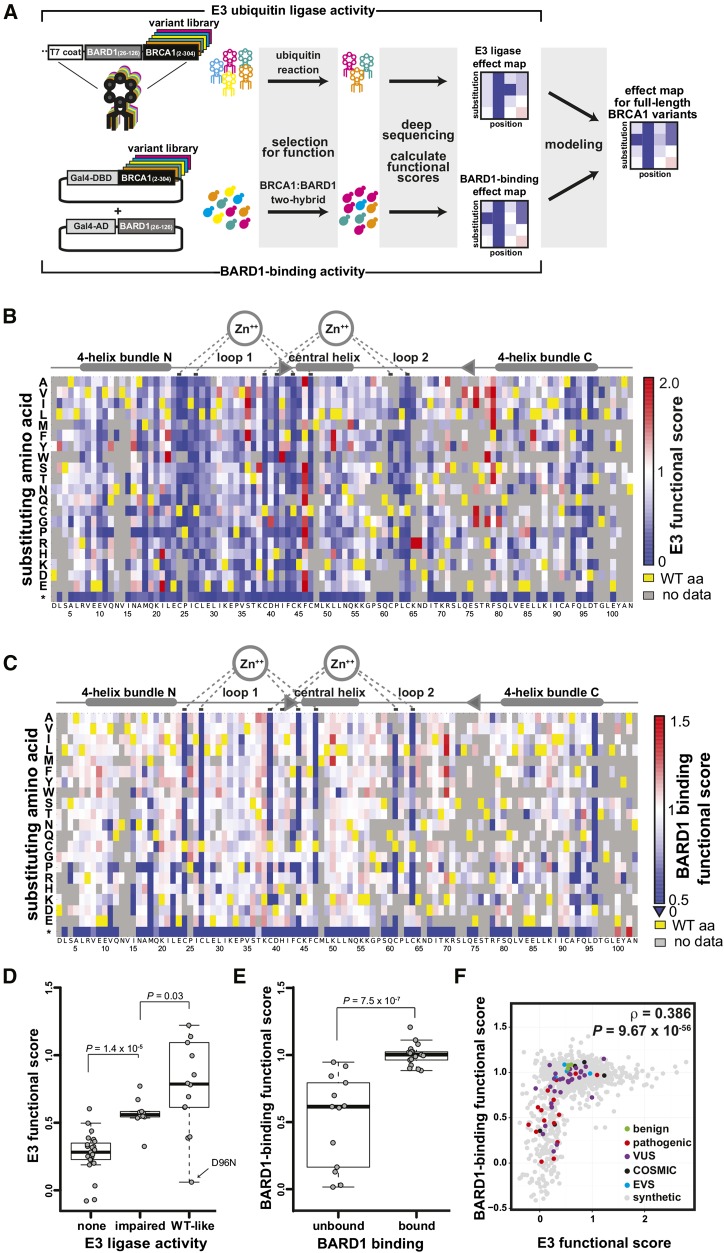
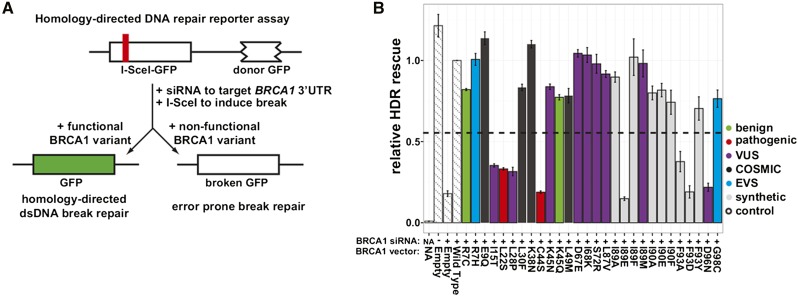
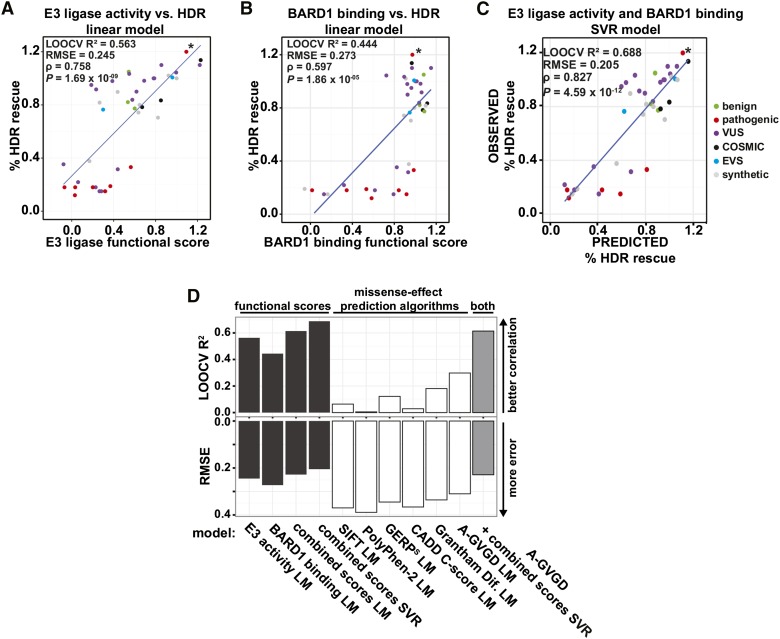
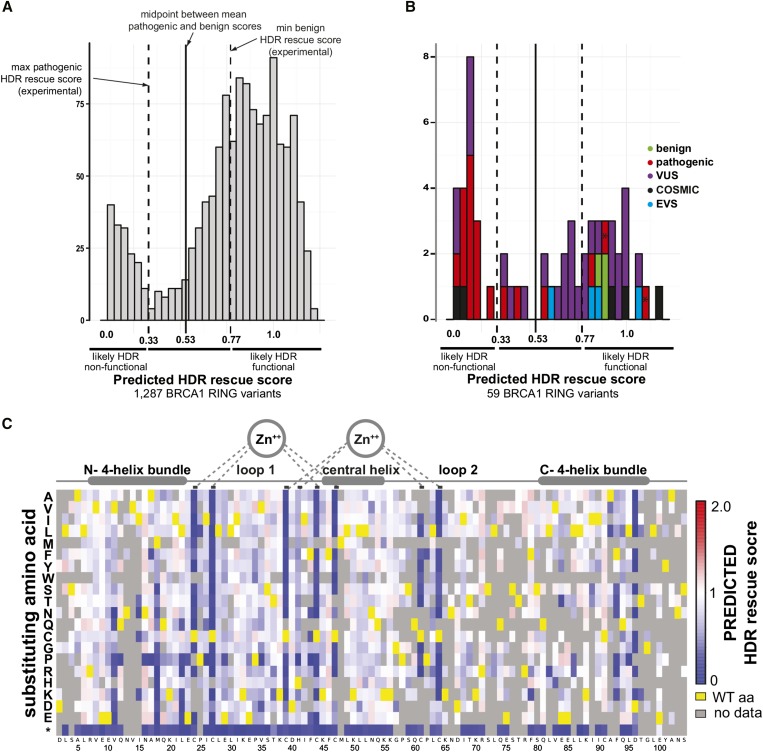
Interpreting variants of uncertain significance (VUS) is a central challenge in medical genetics. One approach is to experimentally measure the functional consequences of VUS, but to date this approach has been post hoc and low throughput. Here we use massively parallel assays to measure the effects of nearly 2000 missense substitutions in the RING domain of BRCA1 on its E3 ubiquitin ligase activity and its binding to the BARD1 RING domain. From the resulting scores, we generate a model to predict the capacities of full-length BRCA1 variants to support homology-directed DNA repair, the essential role of BRCA1 in tumor suppression, and show that it outperforms widely used biological-effect prediction algorithms. We envision that massively parallel functional assays may facilitate the prospective interpretation of variants observed in clinical sequencing.
Keywords: BRCA1; deep mutational scanning; human genetic variation; protein function; variants of uncertain significance.
Copyright © 2015 by the Genetics Society of America.
Figures
References
-
- Bouwman P., van der Gulden H., van der Heijden I., Drost R., Klijn C. N., et al. , 2013. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 3: 1142–1155. - PubMed
-
- Brzovic P. S., Rajagopal P., Hoyt D. W., King M. C., Klevit R. E., 2001. Structure of a BRCA1–BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 8: 833–837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
