

A biopsychosocial model for the management of patients with sickle-cell disease transitioning to adult medical care
- PMID: 25832469
- PMCID: PMC4415939
- DOI: 10.1007/s12325-015-0197-1
A biopsychosocial model for the management of patients with sickle-cell disease transitioning to adult medical care
Abstract
The lifespan of patients with sickle-cell disease (SCD) continues to increase, and most affected individuals in high-resource countries now live into adulthood. This necessitates a successful transition from pediatric to adult health care. Care for transitioning patients with SCD often falls to primary care providers who may not be fully aware of the many challenges and issues faced by patients and the current management strategies for SCD. In this review, we aim to close the knowledge gap between primary care providers and specialists who treat transitioning patients with SCD. We describe the challenges and issues encountered by these patients, and we propose a biopsychosocial multidisciplinary approach to the management of the identified issues. Examples of this approach, such as transition-focused integrated care models and quality improvement collaboratives, with the potential to improve health outcomes in adulthood are also described.
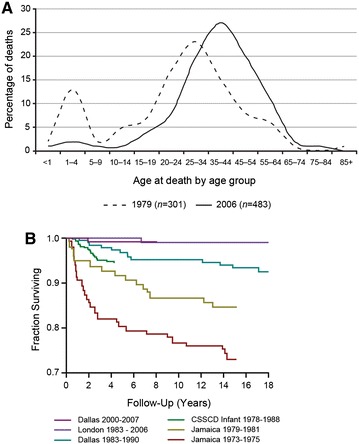
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
