

Maximum-Likelihood Phylogenetic Inference with Selection on Protein Folding Stability
- PMID: 25837579
- PMCID: PMC4833071
- DOI: 10.1093/molbev/msv085
Maximum-Likelihood Phylogenetic Inference with Selection on Protein Folding Stability
Abstract
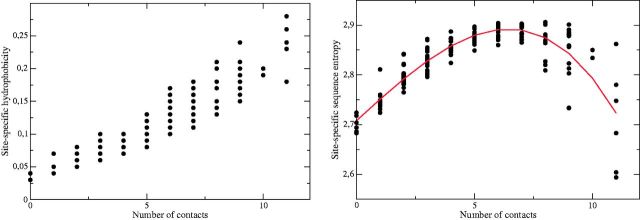
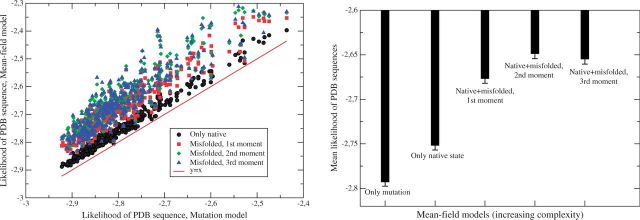
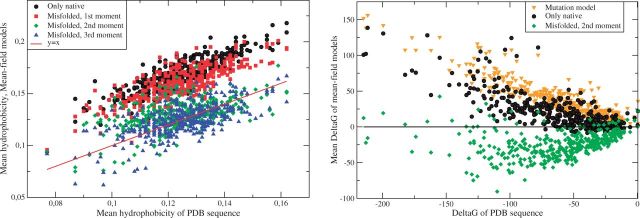
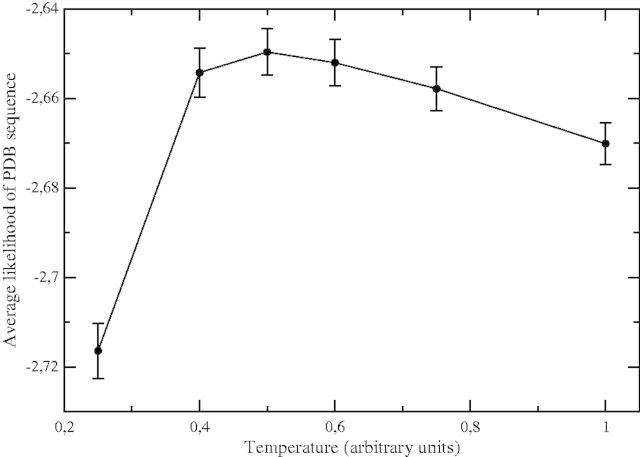
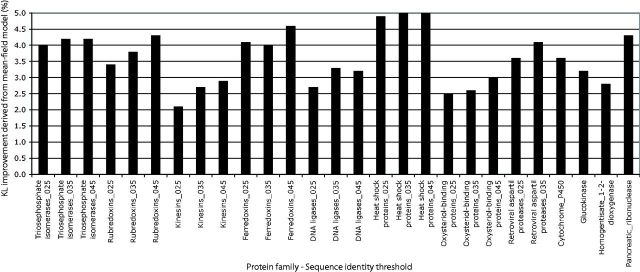
Despite intense work, incorporating constraints on protein native structures into the mathematical models of molecular evolution remains difficult, because most models and programs assume that protein sites evolve independently, whereas protein stability is maintained by interactions between sites. Here, we address this problem by developing a new mean-field substitution model that generates independent site-specific amino acid distributions with constraints on the stability of the native state against both unfolding and misfolding. The model depends on a background distribution of amino acids and one selection parameter that we fix maximizing the likelihood of the observed protein sequence. The analytic solution of the model shows that the main determinant of the site-specific distributions is the number of native contacts of the site and that the most variable sites are those with an intermediate number of native contacts. The mean-field models obtained, taking into account misfolded conformations, yield larger likelihood than models that only consider the native state, because their average hydrophobicity is more realistic, and they produce on the average stable sequences for most proteins. We evaluated the mean-field model with respect to empirical substitution models on 12 test data sets of different protein families. In all cases, the observed site-specific sequence profiles presented smaller Kullback-Leibler divergence from the mean-field distributions than from the empirical substitution model. Next, we obtained substitution rates combining the mean-field frequencies with an empirical substitution model. The resulting mean-field substitution model assigns larger likelihood than the empirical model to all studied families when we consider sequences with identity larger than 0.35, plausibly a condition that enforces conservation of the native structure across the family. We found that the mean-field model performs better than other structurally constrained models with similar or higher complexity. With respect to the much more complex model recently developed by Bordner and Mittelmann, which takes into account pairwise terms in the amino acid distributions and also optimizes the exchangeability matrix, our model performed worse for data with small sequence divergence but better for data with larger sequence divergence. The mean-field model has been implemented into the computer program Prot_Evol that is freely available at http://ub.cbm.uam.es/software/Prot_Evol.php.
Keywords: folding stability; maximum-likelihood estimate; misfolded state; structurally constrained substitution models.
© The Author 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Akaike H. A new look at the statistical model identification. IEEE Trans Automatic Control. 1974;19:716–723.
-
- Babajide A, Hofacker IL, Sippl MJ, Stadler PF. Neutral networks in protein space: a computational study based on knowledge-based potentials of mean force. Fold Des. 1997;2:261–269. - PubMed
-
- Bastolla U, Farwer J, Knapp EW, Vendruscolo M. How to guarantee optimal stability for most representative structures in the Protein Data Bank. Proteins. 2001;44:79–96. - PubMed
-
- Bastolla U, Moya A, Viguera E, van Ham RC. Genomic determinants of protein folding thermodynamics in prokaryotic organisms. J Mol Biol. 2004;343:1451–1466. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
