

Deconvolving the recognition of DNA shape from sequence
- PMID: 25843630
- PMCID: PMC4422406
- DOI: 10.1016/j.cell.2015.02.008
Deconvolving the recognition of DNA shape from sequence
Abstract
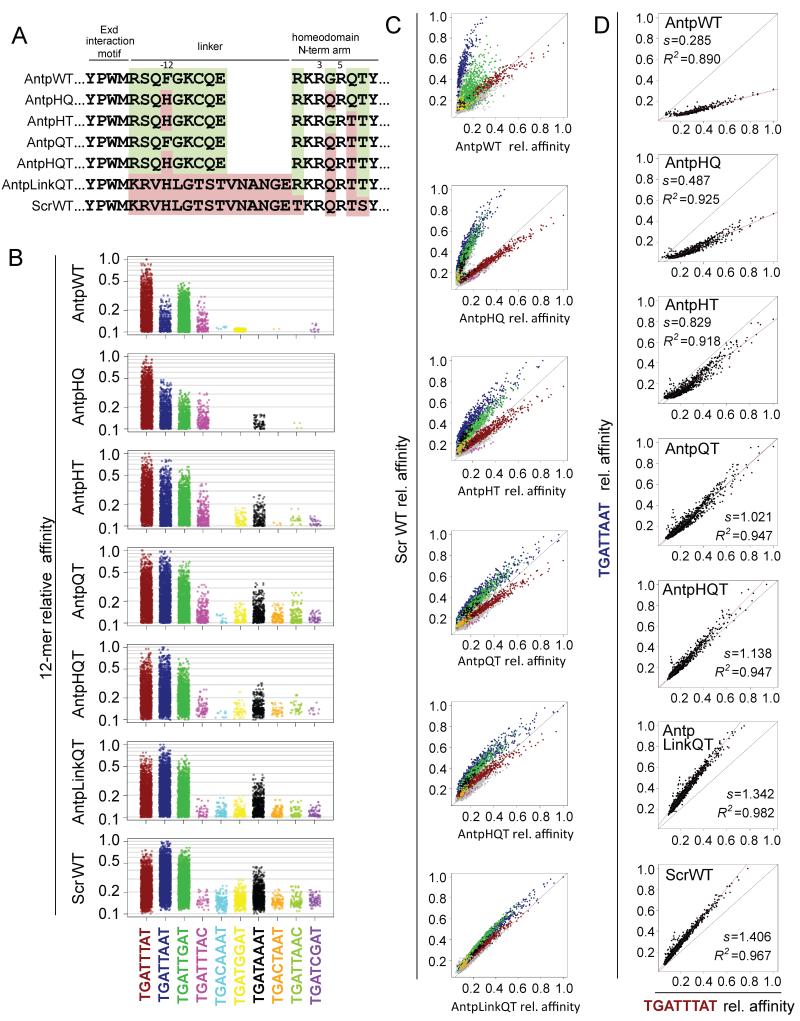
Protein-DNA binding is mediated by the recognition of the chemical signatures of the DNA bases and the 3D shape of the DNA molecule. Because DNA shape is a consequence of sequence, it is difficult to dissociate these modes of recognition. Here, we tease them apart in the context of Hox-DNA binding by mutating residues that, in a co-crystal structure, only recognize DNA shape. Complexes made with these mutants lose the preference to bind sequences with specific DNA shape features. Introducing shape-recognizing residues from one Hox protein to another swapped binding specificities in vitro and gene regulation in vivo. Statistical machine learning revealed that the accuracy of binding specificity predictions improves by adding shape features to a model that only depends on sequence, and feature selection identified shape features important for recognition. Thus, shape readout is a direct and independent component of binding site selection by Hox proteins.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
- R01 GM058575/GM/NIGMS NIH HHS/United States
- R01 HG003008/HG/NHGRI NIH HHS/United States
- R01GM106056/GM/NIGMS NIH HHS/United States
- U01 GM103804/GM/NIGMS NIH HHS/United States
- R35 GM118336/GM/NIGMS NIH HHS/United States
- U01GM103804/GM/NIGMS NIH HHS/United States
- R01 GM054510/GM/NIGMS NIH HHS/United States
- R01 GM106056/GM/NIGMS NIH HHS/United States
- F32 GM099160/GM/NIGMS NIH HHS/United States
- F32GM099160/GM/NIGMS NIH HHS/United States
- R01HG003008/HG/NHGRI NIH HHS/United States
- R01GM058575/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
