

NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer
- PMID: 25843712
- PMCID: PMC4394036
- DOI: 10.1016/j.celrep.2015.03.012
NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer
Abstract
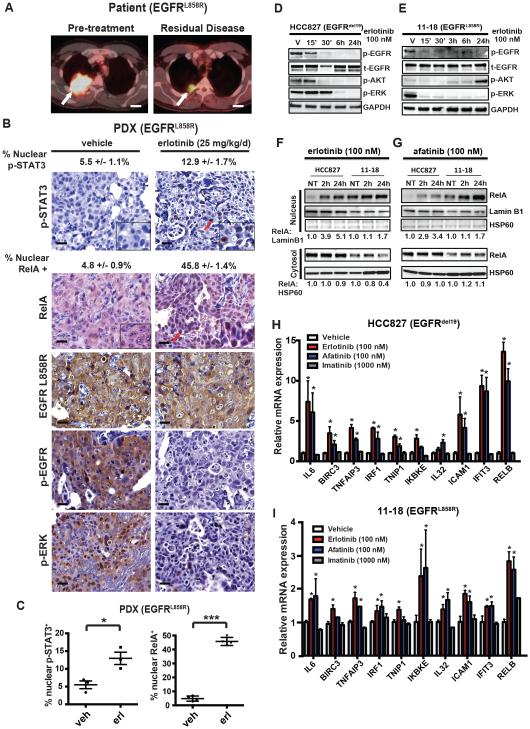
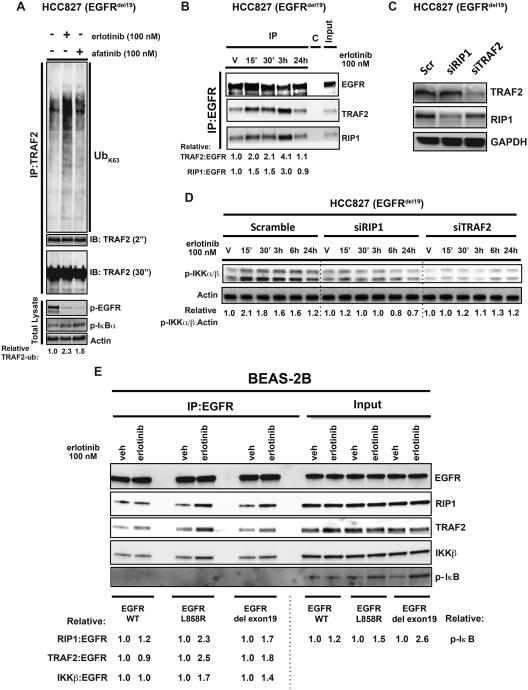
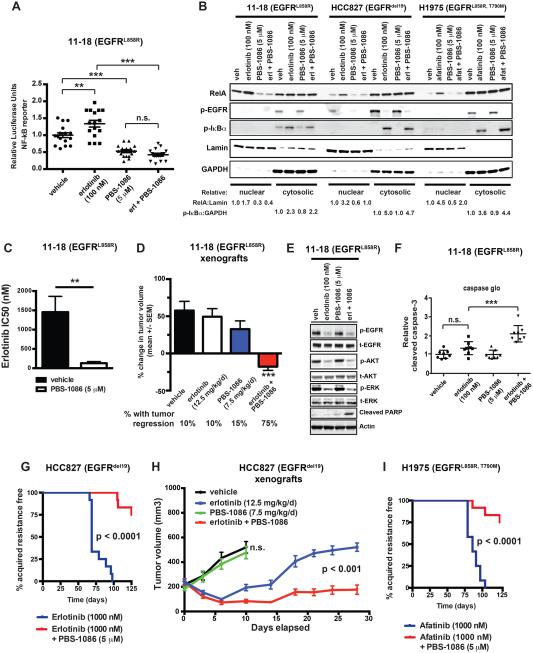
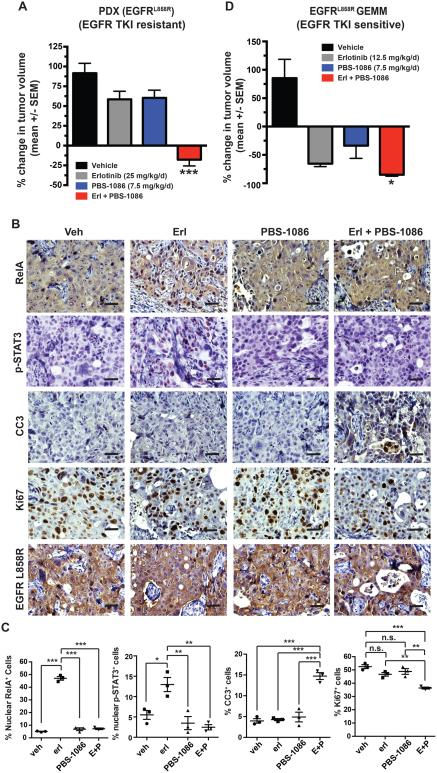
Although oncogene-targeted therapy often elicits profound initial tumor responses in patients, responses are generally incomplete because some tumor cells survive initial therapy as residual disease that enables eventual acquired resistance. The mechanisms underlying tumor cell adaptation and survival during initial therapy are incompletely understood. Here, through the study of EGFR mutant lung adenocarcinoma, we show that NF-κB signaling is rapidly engaged upon initial EGFR inhibitor treatment to promote tumor cell survival and residual disease. EGFR oncogene inhibition induced an EGFR-TRAF2-RIP1-IKK complex that stimulated an NF-κB-mediated transcriptional survival program. The direct NF-κB inhibitor PBS-1086 suppressed this adaptive survival program and increased the magnitude and duration of initial EGFR inhibitor response in multiple NSCLC models, including a patient-derived xenograft. These findings unveil NF-κB activation as a critical adaptive survival mechanism engaged by EGFR oncogene inhibition and provide rationale for EGFR and NF-κB co-inhibition to eliminate residual disease and enhance patient responses.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Aoki Y, Kao PN. Cyclosporin A-sensitive calcium signaling represses NFkappaB activation in human bronchial epithelial cells and enhances NFkappaB activation in Jurkat T-cells. Biochem Biophys Res Commun. 1997;234:424–431. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
