

Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy
- PMID: 25844556
- PMCID: PMC4944827
- DOI: 10.1001/jamaneurol.2015.0178
Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy
Erratum in
-
Second Author Affiliation Missing. Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy.JAMA Neurol. 2015 Oct;72(10):1210. doi: 10.1001/jamaneurol.2015.2916. JAMA Neurol. 2015. PMID: 26457638 No abstract available.
Abstract
Importance: The important depletion of mitochondrial DNA (mtDNA) and the general depression of mitochondrial respiratory chain complex levels (including complex II) have been confirmed, implying an increasing paucity of mitochondria in the muscle from patients with types I, II, and III spinal muscular atrophy (SMA-I, -II, and -III, respectively).
Objective: To investigate mitochondrial dysfunction in a large series of muscle biopsy samples from patients with SMA.
Design, setting, and participants: We studied quadriceps muscle samples from 24 patients with genetically documented SMA and paraspinal muscle samples from 3 patients with SMA-II undergoing surgery for scoliosis correction. Postmortem muscle samples were obtained from 1 additional patient. Age-matched controls consisted of muscle biopsy specimens from healthy children aged 1 to 3 years who had undergone analysis for suspected myopathy. Analyses were performed at the Neuromuscular Unit, Istituto di Ricovero e Cura a Carattere Scientifico Foundation Ca' Granda Ospedale Maggiore Policlinico-Milano, from April 2011 through January 2015.
Exposures: We used histochemical, biochemical, and molecular techniques to examine the muscle samples.
Main outcomes and measures: Respiratory chain activity and mitochondrial content.
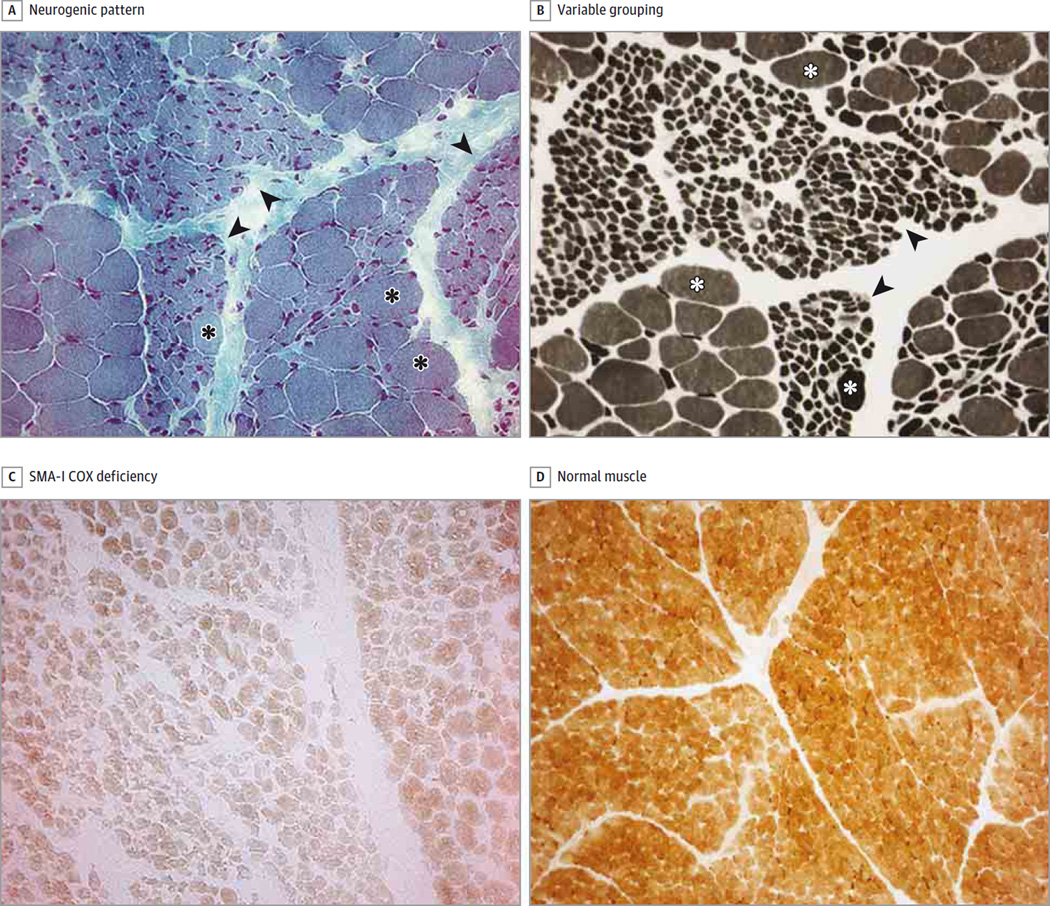
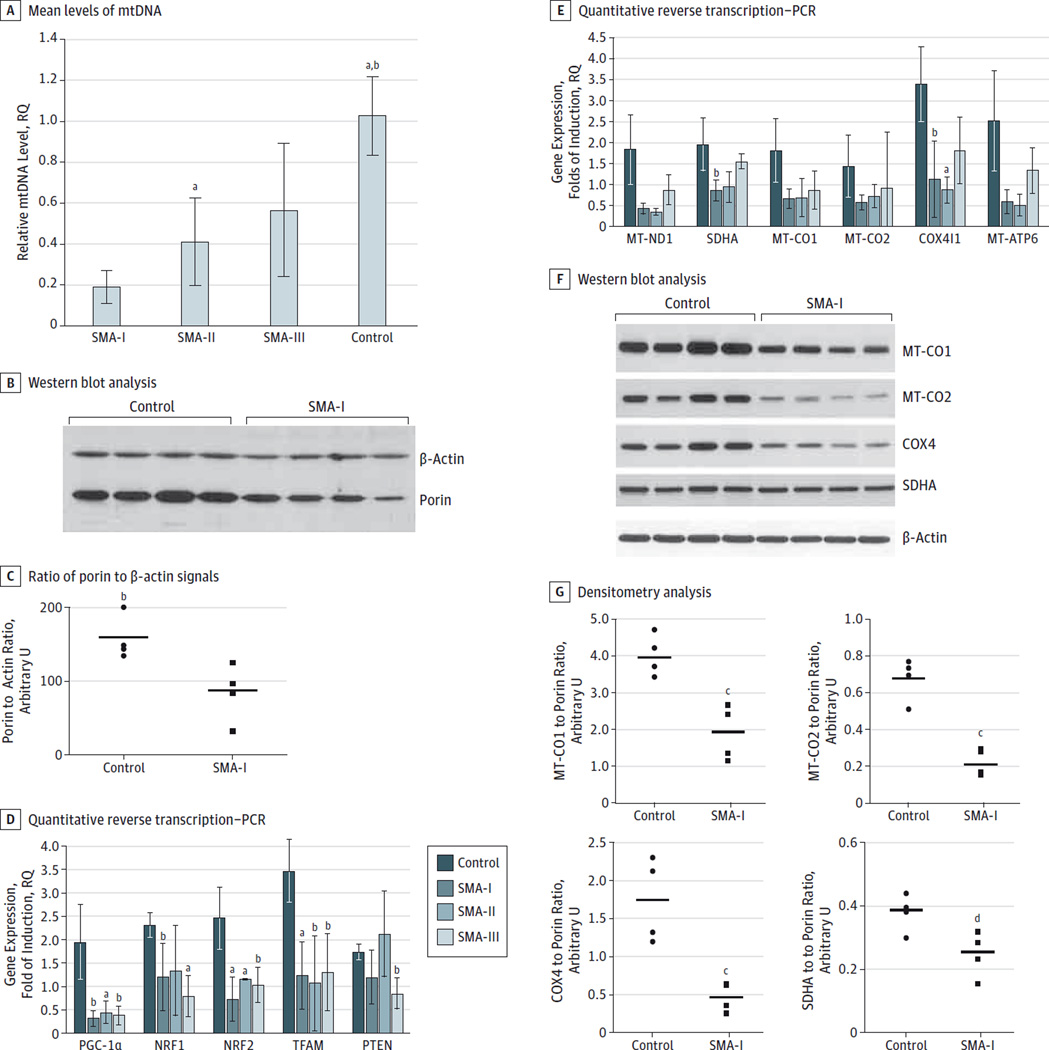
Results: Results of histochemical analysis revealed that cytochrome-c oxidase (COX) deficiency was more evident in muscle samples from patients with SMA-I and SMA-II. Residual activities for complexes I, II, and IV in muscles from patients with SMA-I were 41%, 27%, and 30%, respectively, compared with control samples (P < .005). Muscle mtDNA content and cytrate synthase activity were also reduced in all 3 SMA types (P < .05). We linked these alterations to downregulation of peroxisome proliferator-activated receptor coactivator 1α, the transcriptional activators nuclear respiratory factor 1 and nuclear respiratory factor 2, mitochondrial transcription factor A, and their downstream targets, implying depression of the entire mitochondrial biogenesis. Results of Western blot analysis confirmed the reduced levels of the respiratory chain subunits that included mitochondrially encoded COX1 (47.5%; P = .004), COX2 (32.4%; P < .001), COX4 (26.6%; P < .001), and succinate dehydrogenase complex subunit A (65.8%; P = .03) as well as the structural outer membrane mitochondrial porin (33.1%; P < .001). Conversely, the levels of expression of 3 myogenic regulatory factors-muscle-specific myogenic factor 5, myoblast determination 1, and myogenin-were higher in muscles from patients with SMA compared with muscles from age-matched controls (P < .05).
Conclusions and relevance: Our results strongly support the conclusion that an altered regulation of myogenesis and a downregulated mitochondrial biogenesis contribute to pathologic change in the muscle of patients with SMA. Therapeutic strategies should aim at counteracting these changes.
Conflict of interest statement
Figures


References
-
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–2133. - PubMed
-
- Sossi V, Giuli A, Vitali T, et al. Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur J Hum Genet. 2001;9(2):113–120. - PubMed
-
- Engel AG, Armstrong-Franzini C. Myology. 2nd. New York, NY: McGraw-Hill; 1994.
-
- Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2-13.3. Nature. 1990;344(6266):540–541. - PubMed
-
- Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy–determining gene. Cell. 1995;80(1):155–165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
