

Molecular dynamics simulations of large macromolecular complexes
- PMID: 25845770
- PMCID: PMC4476923
- DOI: 10.1016/j.sbi.2015.03.007
Molecular dynamics simulations of large macromolecular complexes
Abstract
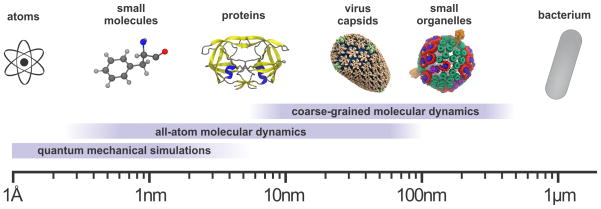
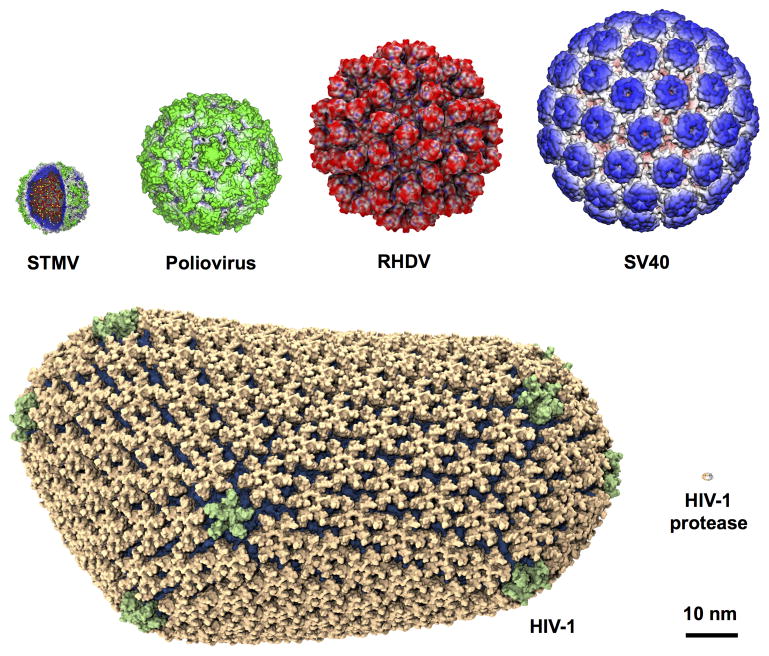
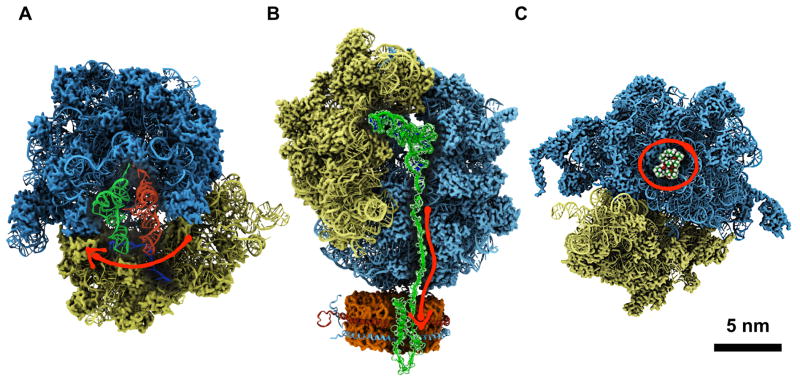
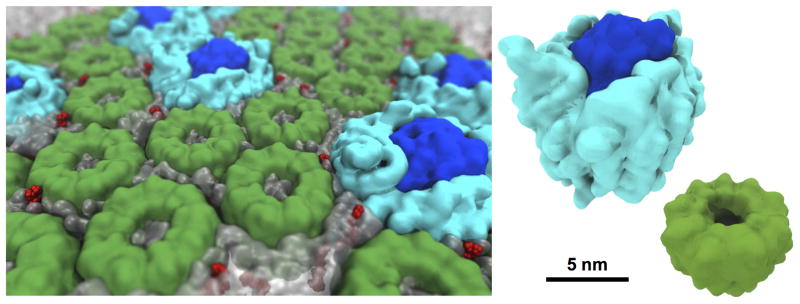
Connecting dynamics to structural data from diverse experimental sources, molecular dynamics simulations permit the exploration of biological phenomena in unparalleled detail. Advances in simulations are moving the atomic resolution descriptions of biological systems into the million-to-billion atom regime, in which numerous cell functions reside. In this opinion, we review the progress, driven by large-scale molecular dynamics simulations, in the study of viruses, ribosomes, bioenergetic systems, and other diverse applications. These examples highlight the utility of molecular dynamics simulations in the critical task of relating atomic detail to the function of supramolecular complexes, a task that cannot be achieved by smaller-scale simulations or existing experimental approaches alone.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
References
-
- Mei C, Sun Y, Zheng G, Bohm EJ, Kalé LV, Phillips JC, Harrison C. Enabling and scaling biomolecular simulations of 100 million atoms on petascale machines with a multicore-optimized message-driven runtime. Proceedings of the 2011 ACM/IEEE conference on Supercomputing; Seattle, WA. 2011.
-
- Roberts E. Cellular and molecular structure as a unifying framework for whole-cell modeling. Current opinion in structural biology. 2014;25:86–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
