

Alström Syndrome: Mutation Spectrum of ALMS1
- PMID: 25846608
- PMCID: PMC4475486
- DOI: 10.1002/humu.22796
Alström Syndrome: Mutation Spectrum of ALMS1
Abstract
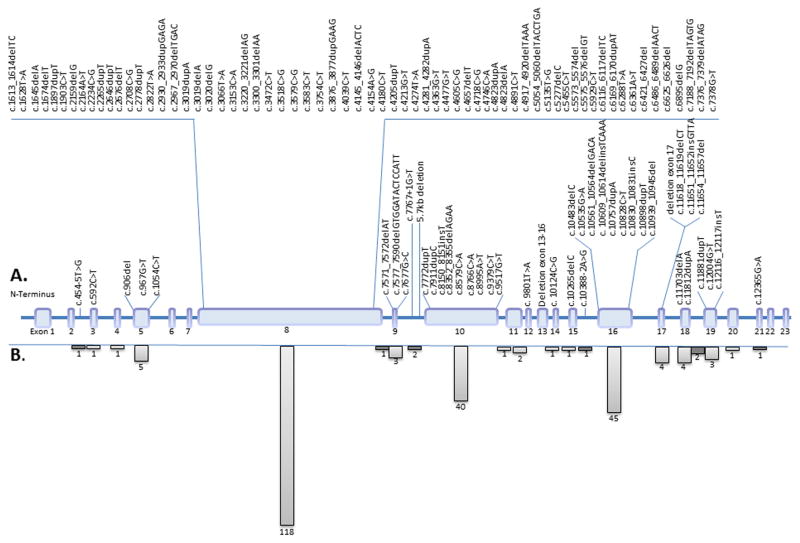
Alström Syndrome (ALMS), a recessive, monogenic ciliopathy caused by mutations in ALMS1, is typically characterized by multisystem involvement including early cone-rod retinal dystrophy and blindness, hearing loss, childhood obesity, type 2 diabetes mellitus, cardiomyopathy, fibrosis, and multiple organ failure. The precise function of ALMS1 remains elusive, but roles in endosomal and ciliary transport and cell cycle regulation have been shown. The aim of our study was to further define the spectrum of ALMS1 mutations in patients with clinical features of ALMS. Mutational analysis in a world-wide cohort of 204 families identified 109 novel mutations, extending the number of known ALMS1 mutations to 239 and highlighting the allelic heterogeneity of this disorder. This study represents the most comprehensive mutation analysis in patients with ALMS, identifying the largest number of novel mutations in a single study worldwide. Here, we also provide an overview of all ALMS1 mutations identified to date.
Keywords: ALMS1; Alström Syndrome; SNV; ciliopathy.
© 2015 WILEY PERIODICALS, INC.
Conflict of interest statement
None of the authors have a conflict of interest.
Figures
References
-
- Aldahmesh MA, Abu-Safieh L, Khan AO, Al-Hassnan ZN, Shaheen R, Rajab M, Monies D, Meyer BF, Alkuraya FS. Allelic heterogeneity in inbred populations: the Saudi experience with Alström syndrome as an illustrative example. Am J Med Genet A. 2009;149(4):662–665. - PubMed
-
- Aliferis K, Hellé S, Gyapay G, Duchatelet S, Stoetzel C, Mandel J-L, Dollfus H. Differentiating Alstrom from Bardet-Biedl syndrome (BBS) using systematic ciliopathy genes sequencing. Ophthalmic Genetics. 2011:1–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
