

Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels
- PMID: 25848093
- PMCID: PMC4468632
- DOI: 10.1124/mol.114.097659
Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels
Abstract
Voltage-gated sodium channels initiate action potentials in nerve, muscle, and other electrically excitable cells. Voltage-gated calcium channels are activated by depolarization during action potentials, and calcium influx through them is the key second messenger of electrical signaling, initiating secretion, contraction, neurotransmission, gene transcription, and many other intracellular processes. Drugs that block sodium channels are used in local anesthesia and the treatment of epilepsy, bipolar disorder, chronic pain, and cardiac arrhythmia. Drugs that block calcium channels are used in the treatment of epilepsy, chronic pain, and cardiovascular disorders, including hypertension, angina pectoris, and cardiac arrhythmia. The principal pore-forming subunits of voltage-gated sodium and calcium channels are structurally related and likely to have evolved from ancestral voltage-gated sodium channels that are widely expressed in prokaryotes. Determination of the structure of a bacterial ancestor of voltage-gated sodium and calcium channels at high resolution now provides a three-dimensional view of the binding sites for drugs acting on sodium and calcium channels. In this minireview, we outline the different classes of sodium and calcium channel drugs, review studies that have identified amino acid residues that are required for their binding and therapeutic actions, and illustrate how the analogs of those key amino acid residues may form drug-binding sites in three-dimensional models derived from bacterial channels.
Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics.
Figures



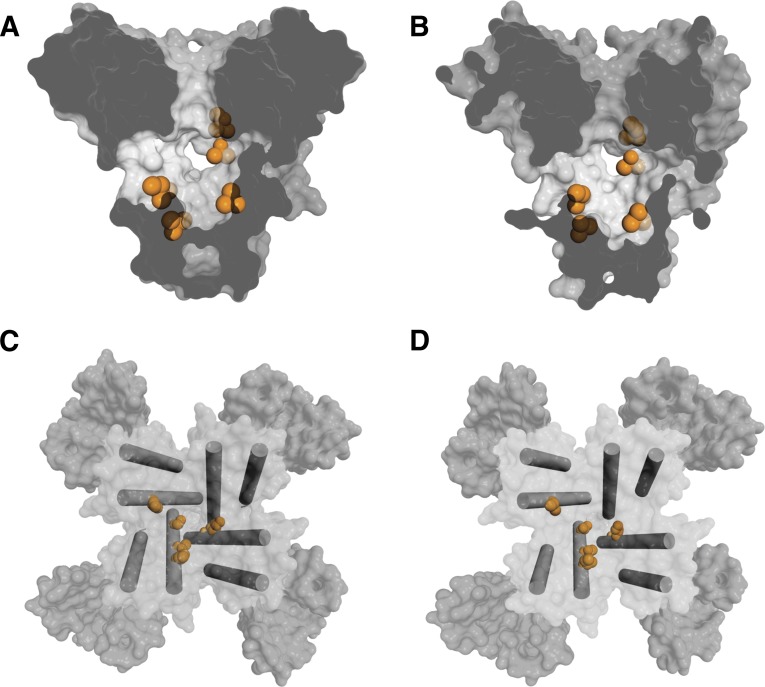

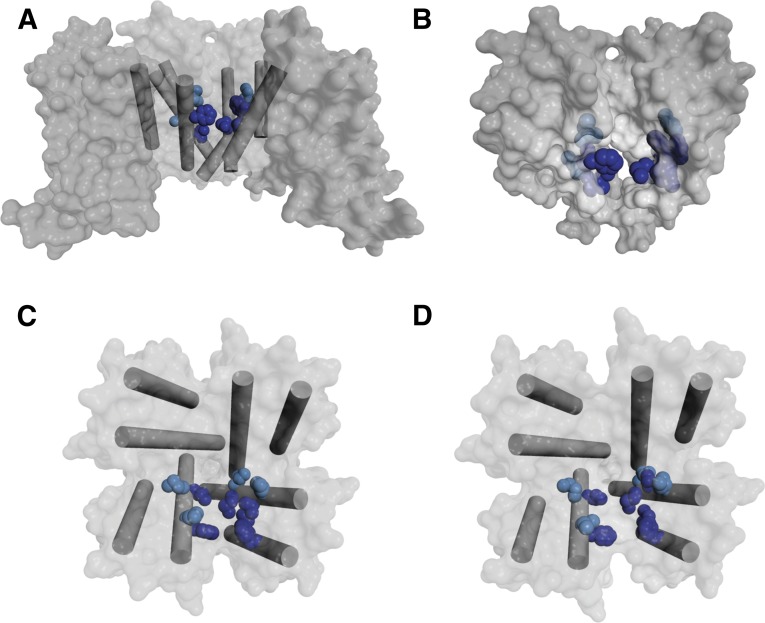
References
-
- Armstrong CM, Bezanilla F. (1973) Currents related to movement of the gating particles of the sodium channels. Nature 242:459–461. - PubMed
-
- Bruhova I, Tikhonov DB, Zhorov BS. (2008) Access and binding of local anesthetics in the closed sodium channel. Mol Pharmacol 74:1033–1045. - PubMed
-
- Catterall WA. (1986) Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem 55:953–985. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
