

Eighteenth-century genomes show that mixed infections were common at time of peak tuberculosis in Europe
- PMID: 25848958
- PMCID: PMC4396363
- DOI: 10.1038/ncomms7717
Eighteenth-century genomes show that mixed infections were common at time of peak tuberculosis in Europe
Abstract
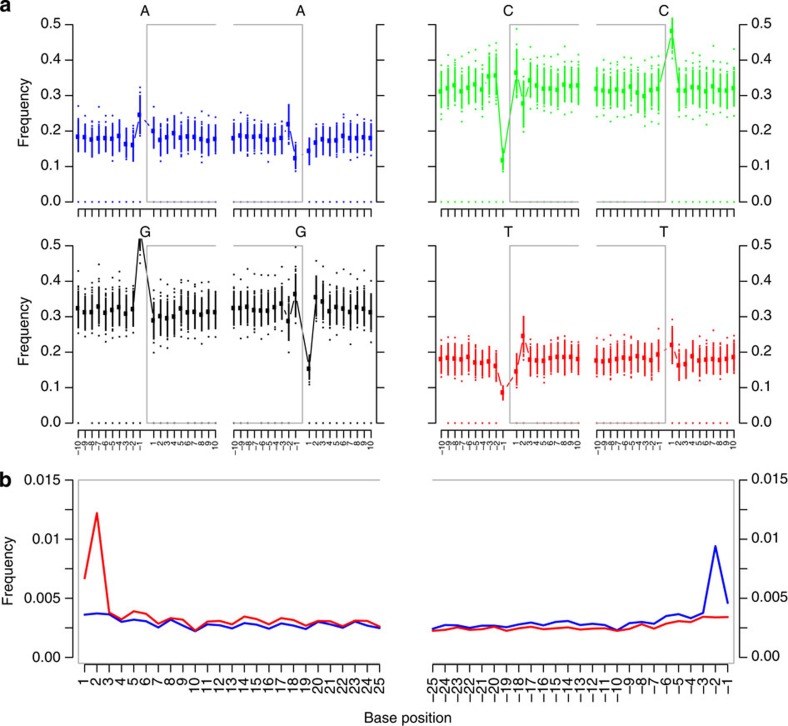
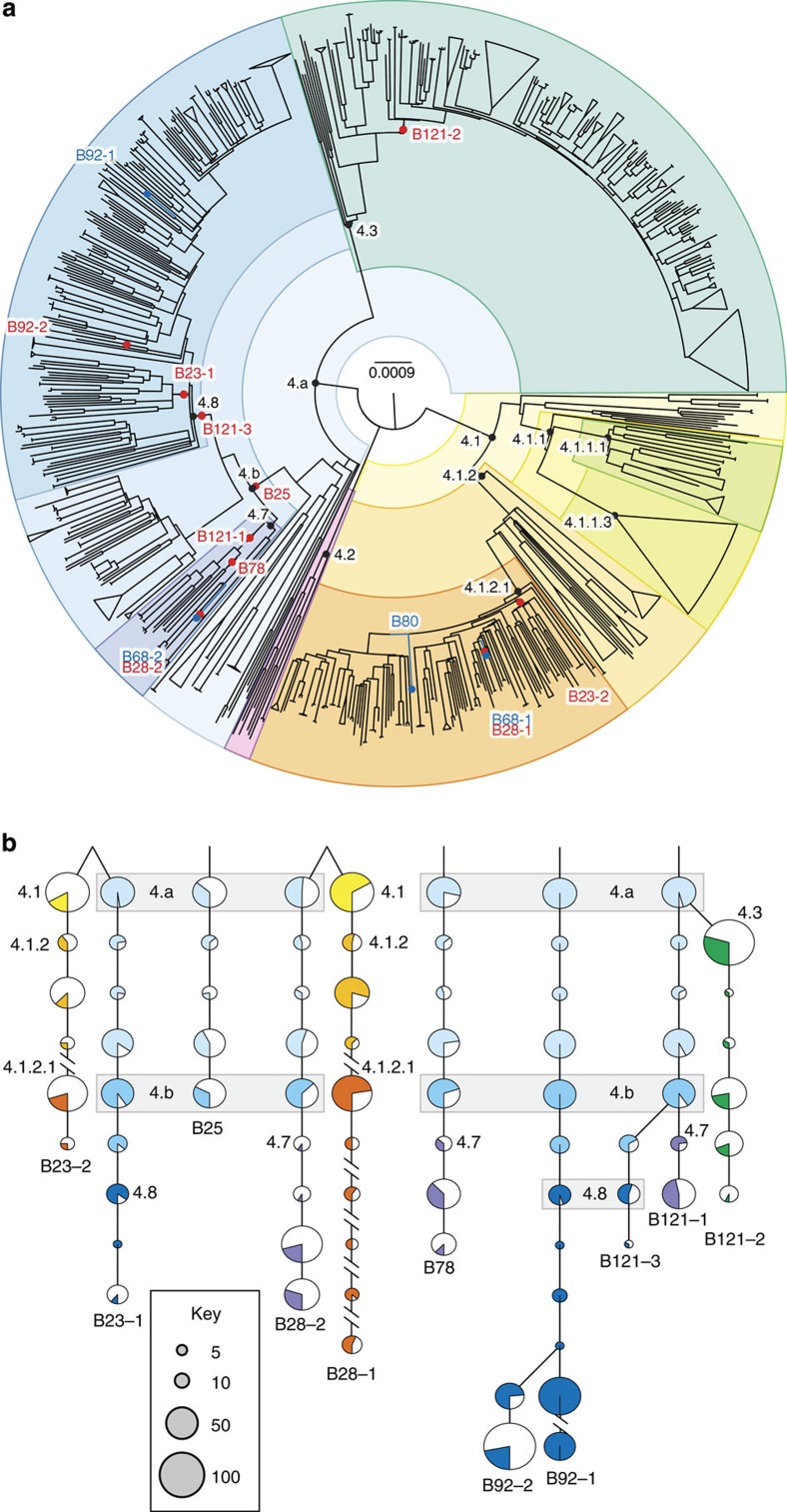
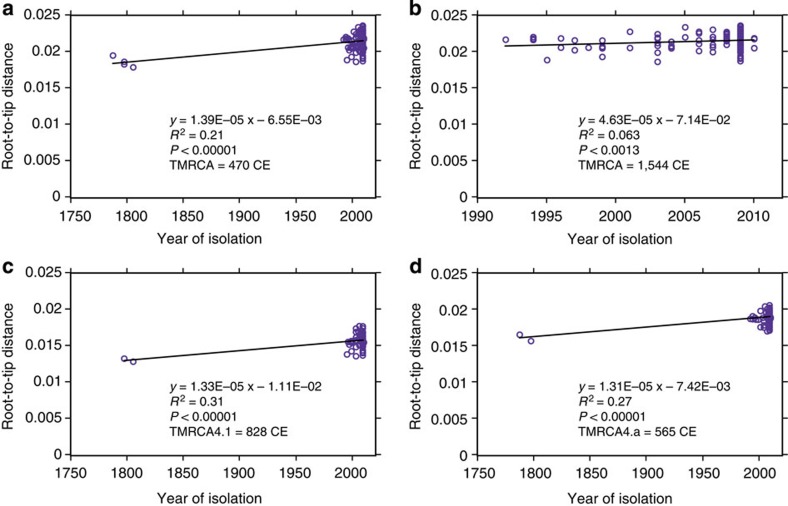
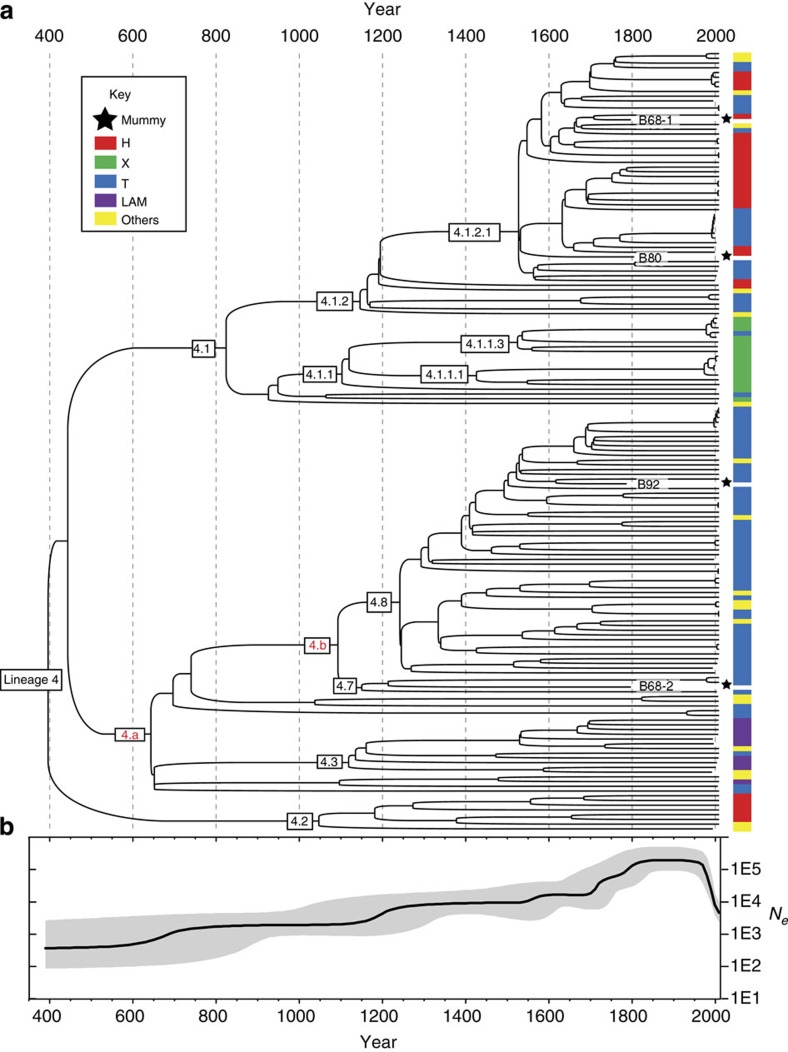
Tuberculosis (TB) was once a major killer in Europe, but it is unclear how the strains and patterns of infection at 'peak TB' relate to what we see today. Here we describe 14 genome sequences of M. tuberculosis, representing 12 distinct genotypes, obtained from human remains from eighteenth-century Hungary using metagenomics. All our historic genotypes belong to M. tuberculosis Lineage 4. Bayesian phylogenetic dating, based on samples with well-documented dates, places the most recent common ancestor of this lineage in the late Roman period. We find that most bodies yielded more than one M. tuberculosis genotype and we document an intimate epidemiological link between infections in two long-dead individuals. Our results suggest that metagenomic approaches usefully inform detection and characterization of historical and contemporary infections.
Figures

References
-
- Koch R. Die Aetiologie der Tuberkulose. Berl. Klin. Wochenschr. 19, 221–230 (1882) .
-
- Donoghue H. D. Insights into ancient leprosy and tuberculosis using metagenomics. Trends Microbiol. 21, 448–450 (2013) . - PubMed
-
- Chan J. Z. et al. Metagenomic analysis of tuberculosis in a mummy. N. Engl. J. Med. 369, 289–290 (2013) . - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
