

GNAS Spectrum of Disorders
- PMID: 25851935
- PMCID: PMC4417430
- DOI: 10.1007/s11914-015-0268-x
GNAS Spectrum of Disorders
Abstract
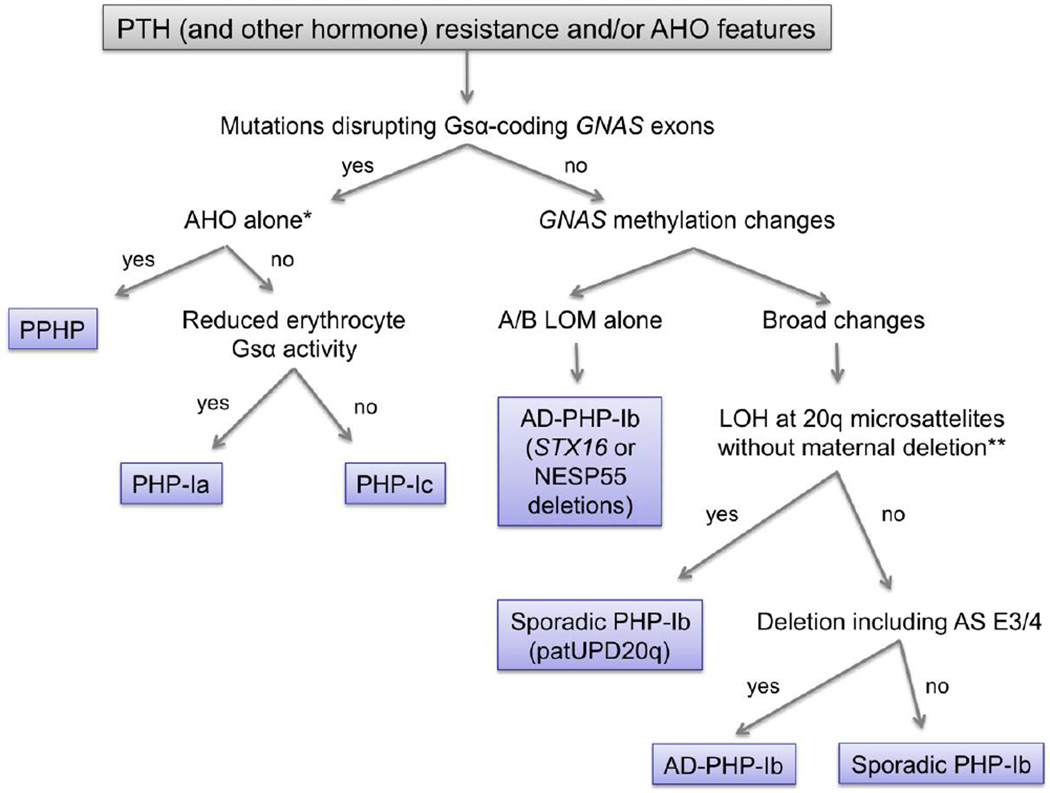
The GNAS complex locus encodes the alpha-subunit of the stimulatory G protein (Gsα), a ubiquitous signaling protein mediating the actions of many hormones, neurotransmitters, and paracrine/autocrine factors via generation of the second messenger cAMP. GNAS gives rise to other gene products, most of which exhibit exclusively monoallelic expression. In contrast, Gsα is expressed biallelically in most tissues; however, paternal Gsα expression is silenced in a small number of tissues through as-yet-poorly understood mechanisms that involve differential methylation within GNAS. Gsα-coding GNAS mutations that lead to diminished Gsα expression and/or function result in Albright's hereditary osteodystrophy (AHO) with or without hormone resistance, i.e., pseudohypoparathyroidism type-Ia/Ic and pseudo-pseudohypoparathyroidism, respectively. Microdeletions that alter GNAS methylation and, thereby, diminish Gsα expression in tissues in which the paternal Gsα allele is normally silenced also cause hormone resistance, which occurs typically in the absence of AHO, a disorder termed pseudohypoparathyroidism type-Ib. Mutations of GNAS that cause constitutive Gsα signaling are found in patients with McCune-Albright syndrome, fibrous dysplasia of bone, and different endocrine and non-endocrine tumors. Clinical features of these diseases depend significantly on the parental allelic origin of the GNAS mutation, reflecting the tissue-specific paternal Gsα silencing. In this article, we review the pathogenesis and the phenotypes of these human diseases.
Conflict of interest statement
S Turan and M Bastepe both declare no conflicts of interest.
Figures


References
-
- Kehlenbach RH, Matthey J, Huttner WB. XL alpha s is a new type of G protein. Nature. 1994;372:804–809. - PubMed
-
- Ischia R, Lovisetti-Scamihorn P, Hogue-Angeletti R, et al. Molecular cloning and characterization of NESP55, a novel chromogranin-like precursor of a peptide with 5-HT1B receptor antagonist activity. J Biol Chem. 1997;272:11657–11662. - PubMed
-
- Ishikawa Y, Bianchi C, Nadal-Ginard B, et al. Alternative promoter and 5’ exon generate a novel Gsα mRNA. J Biol Chem. 1990;265:8458–8462. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
