

Effects of Tumor Necrosis Factor-α on Podocyte Expression of Monocyte Chemoattractant Protein-1 and in Diabetic Nephropathy
- PMID: 25852733
- PMCID: PMC4369122
- DOI: 10.1159/000369576
Effects of Tumor Necrosis Factor-α on Podocyte Expression of Monocyte Chemoattractant Protein-1 and in Diabetic Nephropathy
Abstract
Background/aims: Tumor necrosis factor (TNF)-α is believed to play a role in diabetic kidney disease. This study explores the specific effects of TNF-α with regard to nephropathy-relevant parameters in the podocyte.
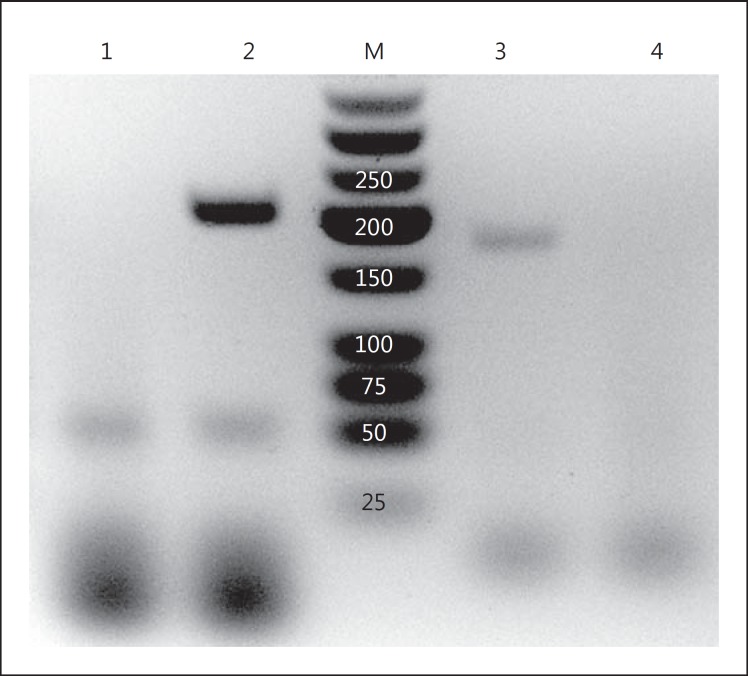
Methods: Cultured mouse podocytes were treated with recombinant TNF-α and assayed for production of monocyte chemoattractant protein-1 (MCP-1) by enzyme-linked immunosorbent assay (ELISA). TNF-α signaling of MCP-1 was elucidated by antibodies against TNF receptor (TNFR) 1 or TNFR2 or inhibitors of nuclear factor-kappaB (NF-κB), phosphatidylinositol 3-kinase (PI3K) or Akt. In vivo studies were done on male db/m and type 2 diabetic db/db mice. Levels of TNF-α and MCP-1 were measured by RT-qPCR and ELISA in the urine, kidney and plasma of the two cohorts and correlated with albuminuria.
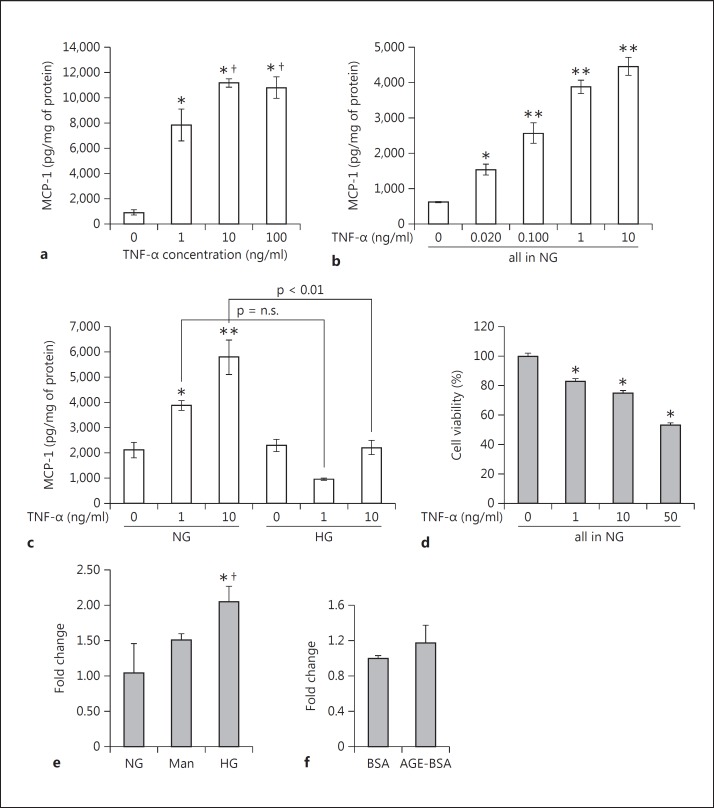
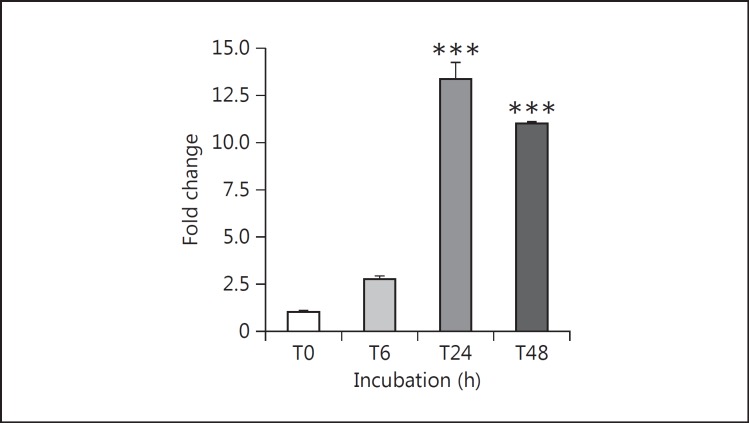
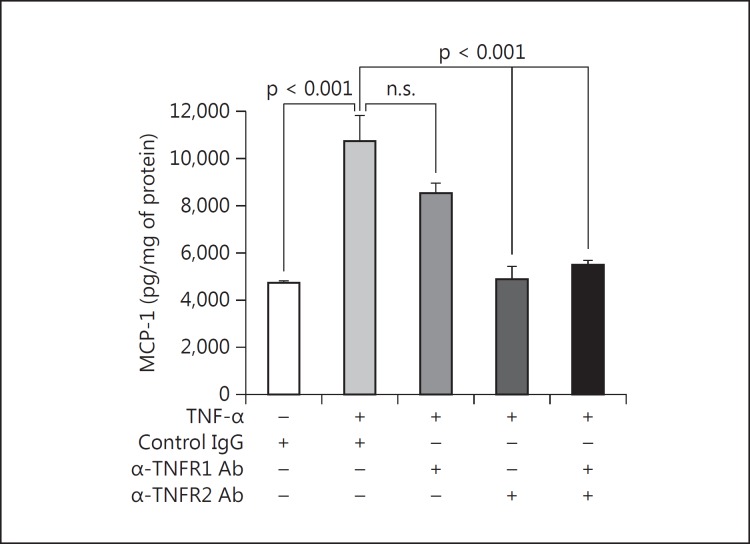
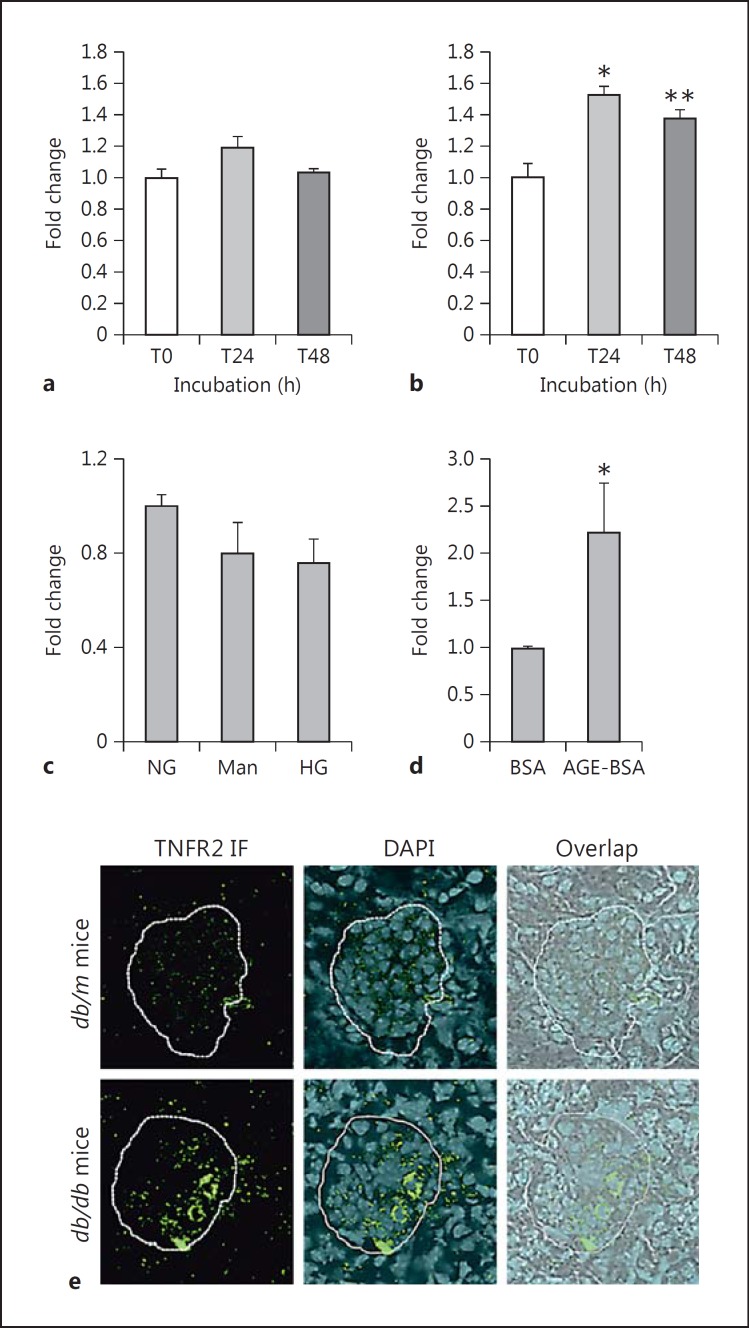
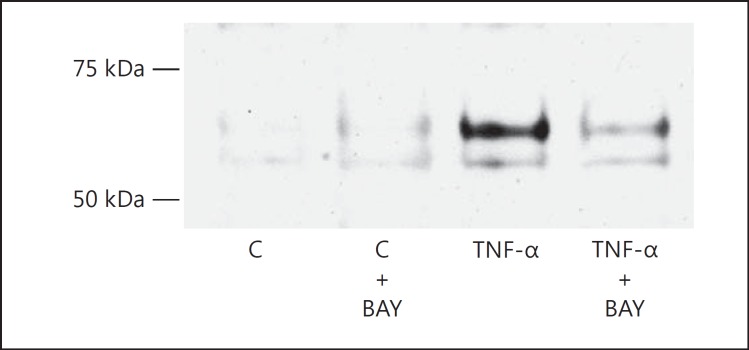
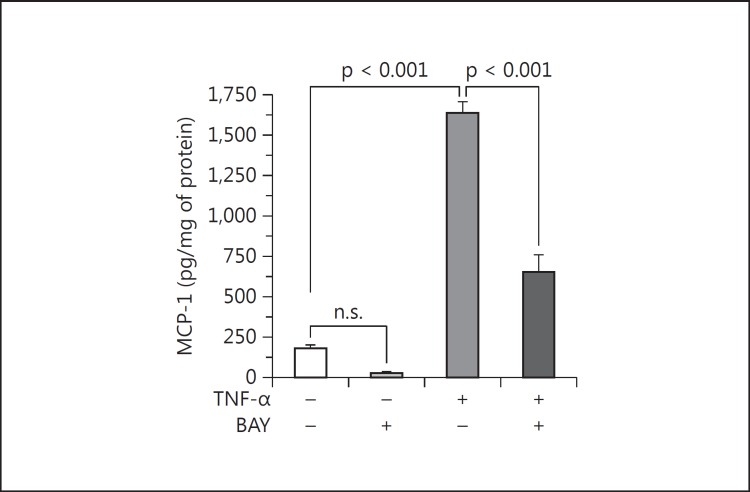
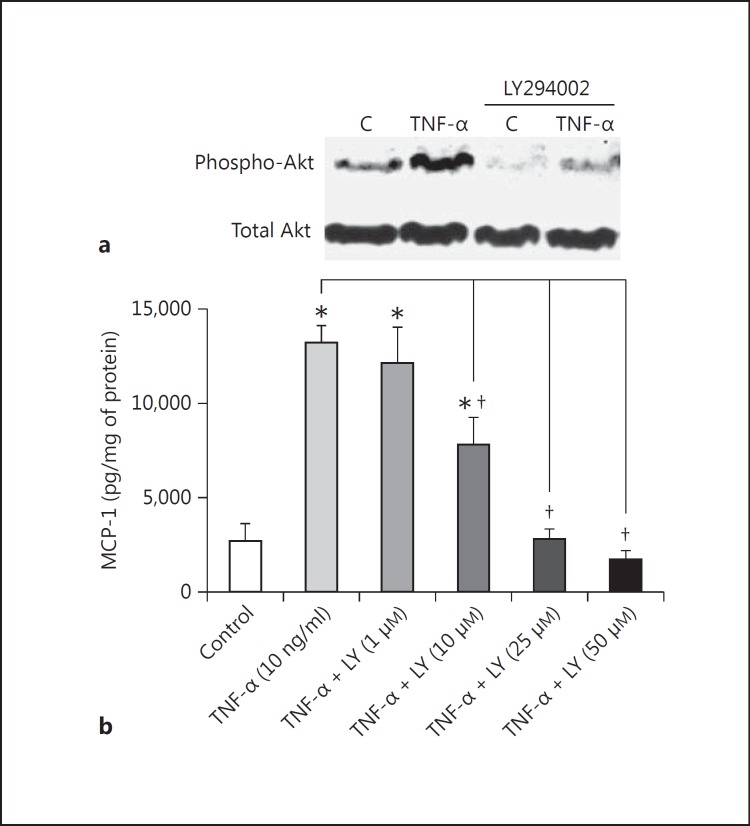
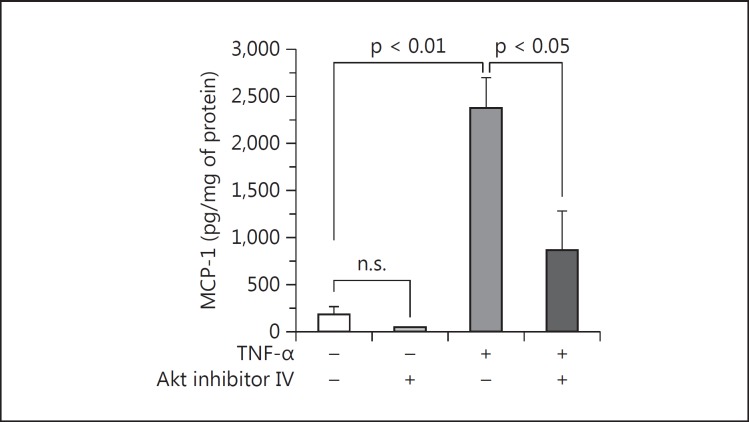
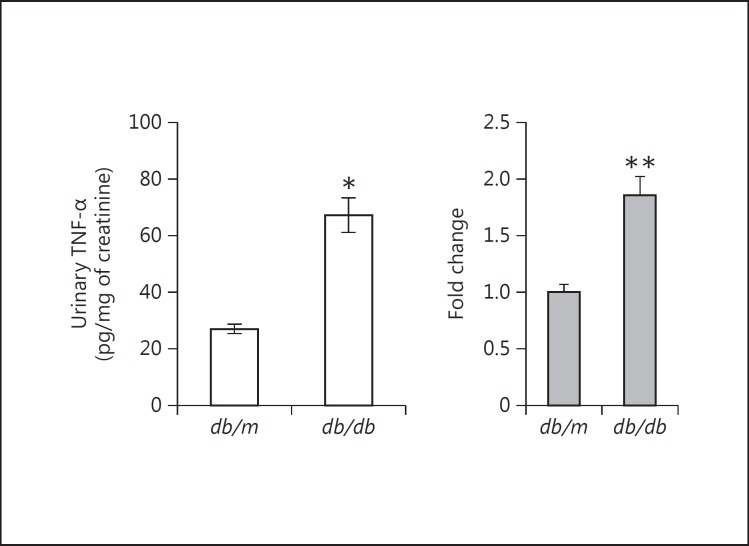
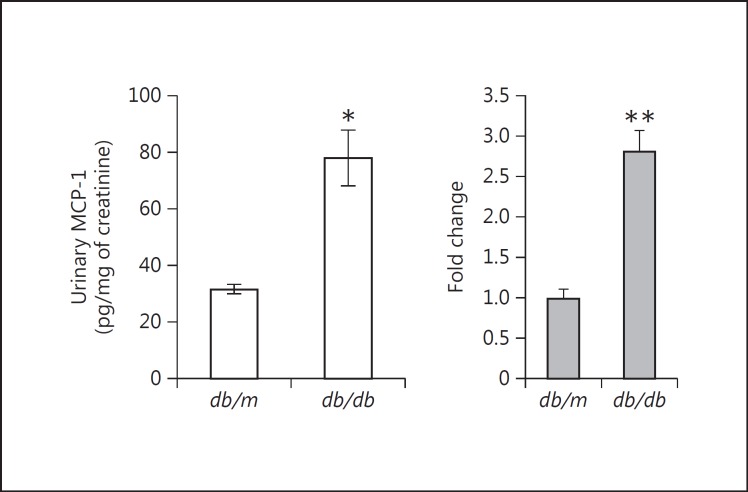
Results: Podocytes treated with TNF-α showed a robust increase (∼900%) in the secretion of MCP-1, induced in a dose- and time-dependent manner. Signaling of MCP-1 expression occurred through TNFR2, which was inducible by TNF-α ligand, but did not depend on TNFR1. TNF-α then proceeded via the NF-κB and the PI3K/Akt systems, based on the effectiveness of the inhibitors of those pathways. For in vivo relevance to diabetic kidney disease, TNF-α and MCP-1 levels were found to be elevated in the urine of db/db mice but not in the plasma.
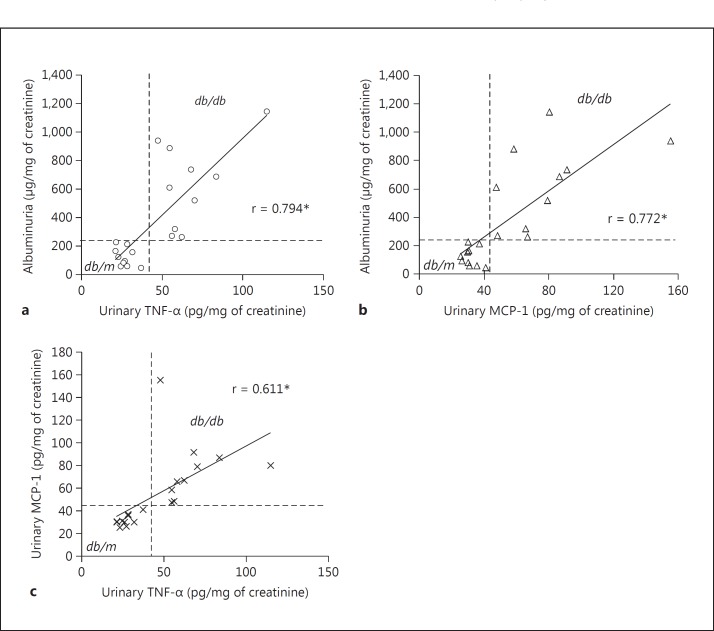
Conclusion: TNF-α potently stimulates podocytes to produce MCP-1, utilizing the TNFR2 receptor and the NF-κB and PI3K/Akt pathways. Both TNF-α and MCP-1 levels were increased in the urine of diabetic db/db mice, correlating with the severity of diabetic albuminuria.
Keywords: Akt or protein kinase B; Albuminuria; Diabetic rodent model; Nuclear factor-kappaB; Phosphatidylinositol 3-kinase; TNF receptor 2.
Figures
References
-
- Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I, Tomino Y, Koide H. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993;42:450–450. - PubMed
-
- Sugimoto H, Shikata K, Wada J, Horiuchi S, Makino H. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia. 1999;42:878–878. - PubMed
-
- Mensah-Brown EP, Obineche EN, Galadari S, Chandranath E, Shahin A, Ahmed I, Patel SM, Adem A. Streptozotocin-induced diabetic nephropathy in rats: the role of inflammatory cytokines. Cytokine. 2005;31:180–180. - PubMed
-
- Navarro JF, Mora C, Muros M, Garcia J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant. 2006;21:3428–3428. - PubMed
-
- Moriwaki Y, Inokuchi T, Yamamoto A, Ka T, Tsutsumi Z, Takahashi S, Yamamoto T. Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol. 2007;44:215–215. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
