

DGEclust: differential expression analysis of clustered count data
- PMID: 25853652
- PMCID: PMC4365804
- DOI: 10.1186/s13059-015-0604-6
DGEclust: differential expression analysis of clustered count data
Abstract
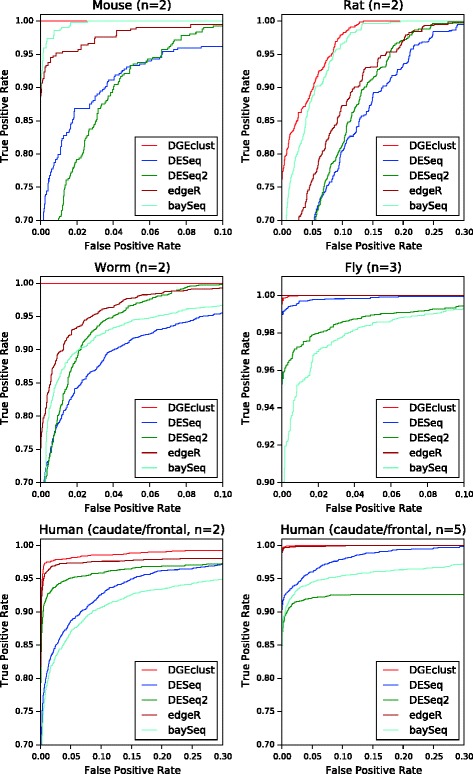
We present a statistical methodology, DGEclust, for differential expression analysis of digital expression data. Our method treats differential expression as a form of clustering, thus unifying these two concepts. Furthermore, it simultaneously addresses the problem of how many clusters are supported by the data and uncertainty in parameter estimation. DGEclust successfully identifies differentially expressed genes under a number of different scenarios, maintaining a low error rate and an excellent control of its false discovery rate with reasonable computational requirements. It is formulated to perform particularly well on low-replicated data and be applicable to multi-group data. DGEclust is available at http://dvav.github.io/dgeclust/.
Figures








Similar articles
-
Probe-level measurement error improves accuracy in detecting differential gene expression.Bioinformatics. 2006 Sep 1;22(17):2107-13. doi: 10.1093/bioinformatics/btl361. Epub 2006 Jul 4. Bioinformatics. 2006. PMID: 16820429
-
Combining Pareto-optimal clusters using supervised learning for identifying co-expressed genes.BMC Bioinformatics. 2009 Jan 20;10:27. doi: 10.1186/1471-2105-10-27. BMC Bioinformatics. 2009. PMID: 19154590 Free PMC article.
-
A computationally efficient modular optimal discovery procedure.Bioinformatics. 2011 Feb 15;27(4):509-15. doi: 10.1093/bioinformatics/btq701. Epub 2010 Dec 24. Bioinformatics. 2011. PMID: 21186247 Free PMC article.
-
How does gene expression clustering work?Nat Biotechnol. 2005 Dec;23(12):1499-501. doi: 10.1038/nbt1205-1499. Nat Biotechnol. 2005. PMID: 16333293 Review.
-
Comparing algorithms for clustering of expression data: how to assess gene clusters.Methods Mol Biol. 2009;541:479-509. doi: 10.1007/978-1-59745-243-4_21. Methods Mol Biol. 2009. PMID: 19381534 Review.
Cited by
-
Hierarchical probabilistic models for multiple gene/variant associations based on next-generation sequencing data.Bioinformatics. 2017 Oct 1;33(19):3058-3064. doi: 10.1093/bioinformatics/btx355. Bioinformatics. 2017. PMID: 28575251 Free PMC article.
-
Eleven grand challenges in single-cell data science.Genome Biol. 2020 Feb 7;21(1):31. doi: 10.1186/s13059-020-1926-6. Genome Biol. 2020. PMID: 32033589 Free PMC article. Review.
-
Scalable nonparametric clustering with unified marker gene selection for single-cell RNA-seq data.bioRxiv [Preprint]. 2024 Feb 12:2024.02.11.579839. doi: 10.1101/2024.02.11.579839. bioRxiv. 2024. PMID: 38405697 Free PMC article. Preprint.
-
A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies.Entropy (Basel). 2019 Mar 4;21(3):242. doi: 10.3390/e21030242. Entropy (Basel). 2019. PMID: 33266957 Free PMC article.
-
MBCdeg4: A modified clustering-based method for identifying differentially expressed genes from RNA-seq data.MethodsX. 2024 Dec 30;14:103149. doi: 10.1016/j.mex.2024.103149. eCollection 2025 Jun. MethodsX. 2024. PMID: 39866202 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
