

α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2
- PMID: 25855184
- PMCID: PMC4388928
- DOI: 10.1523/JNEUROSCI.0632-14.2015
α-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2
Abstract
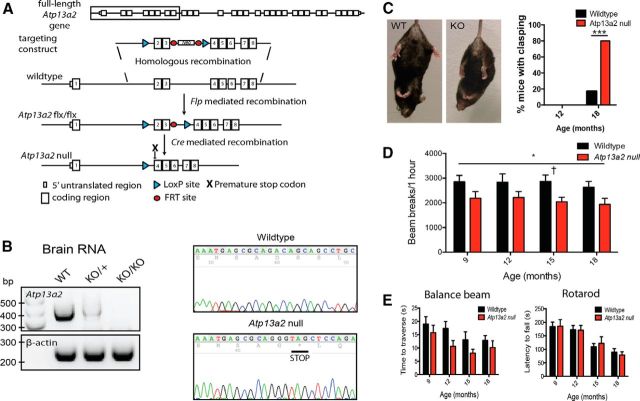
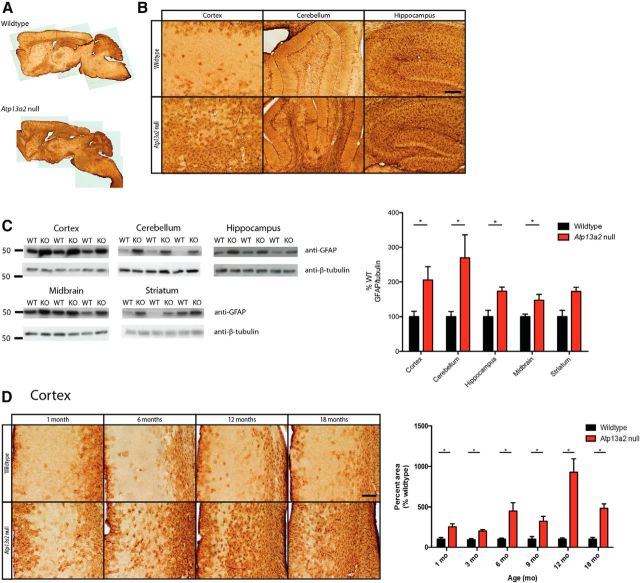
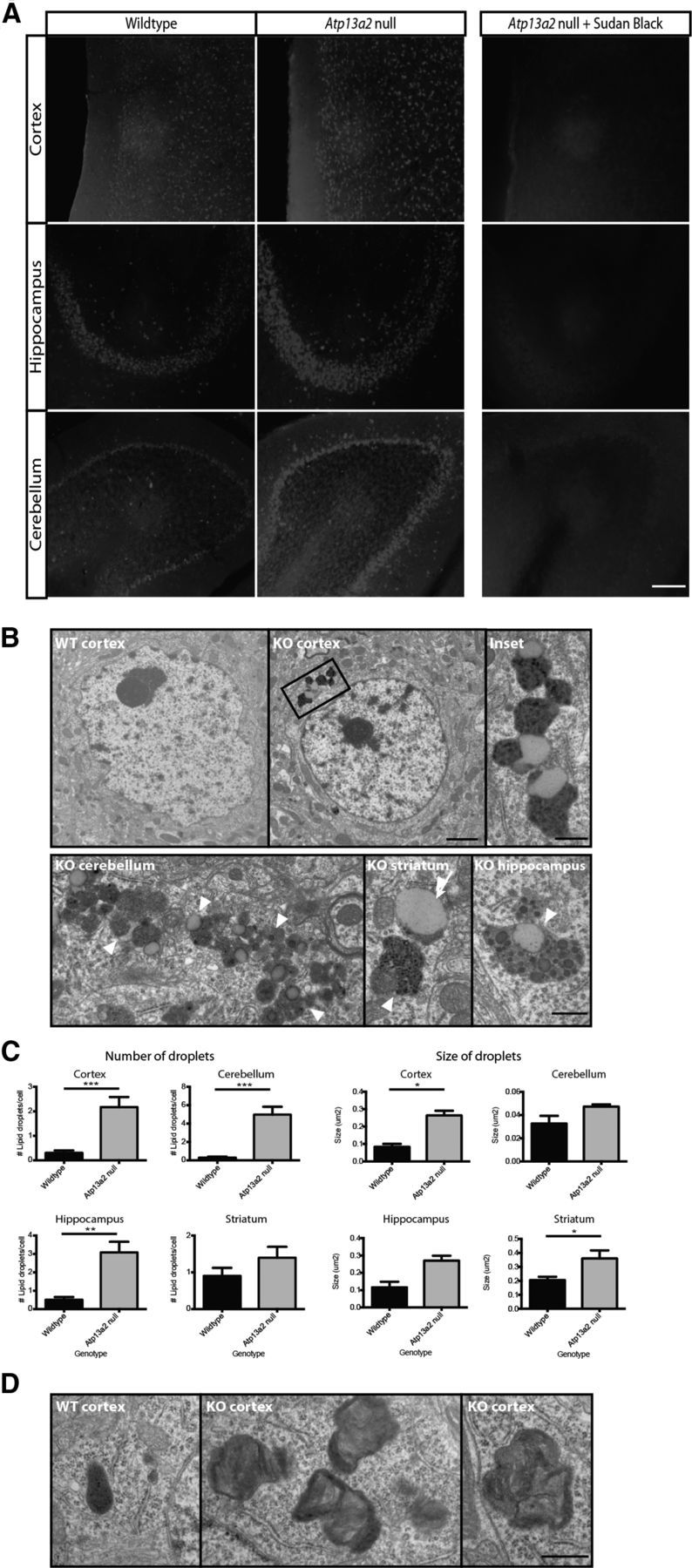
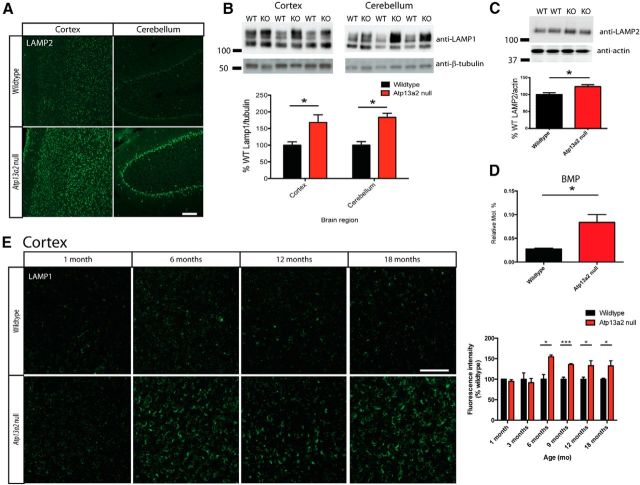
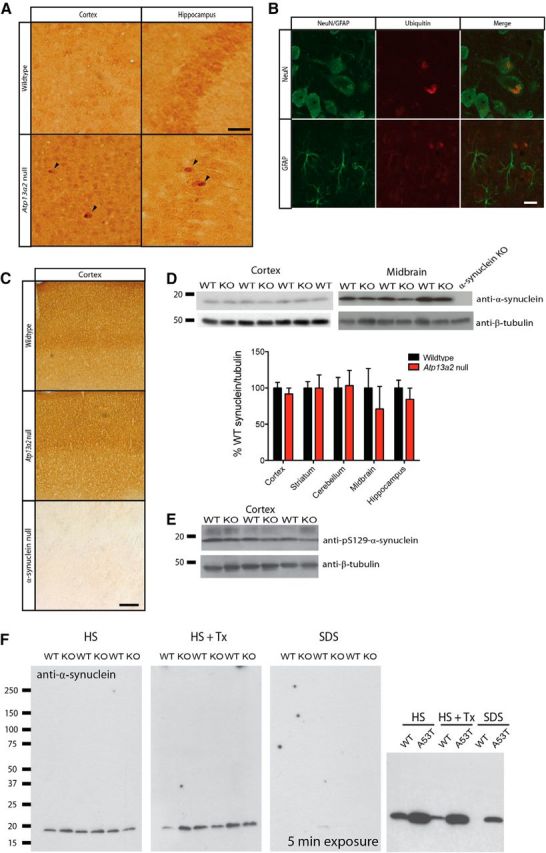
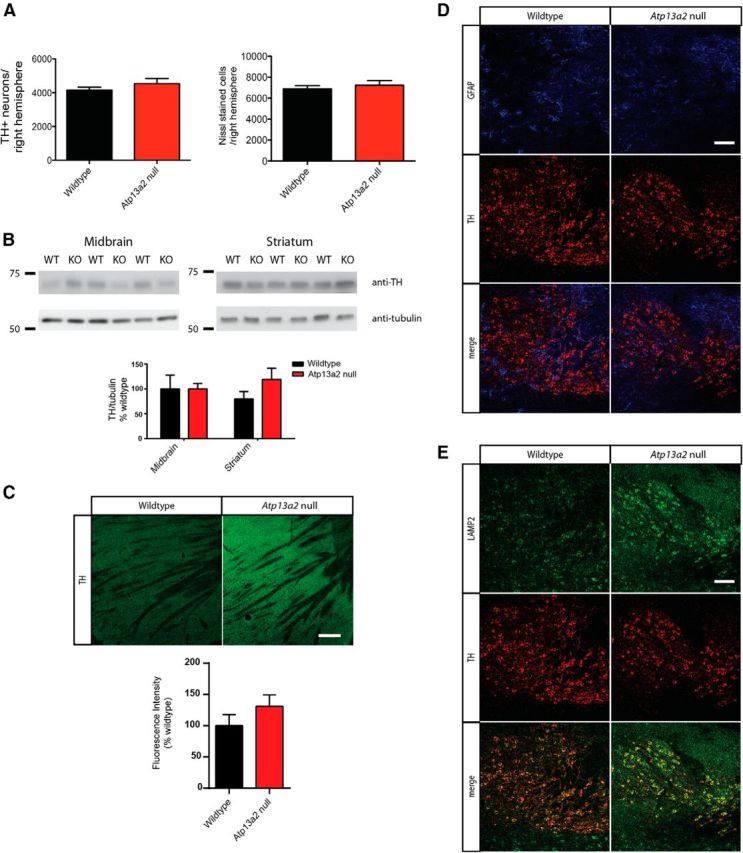
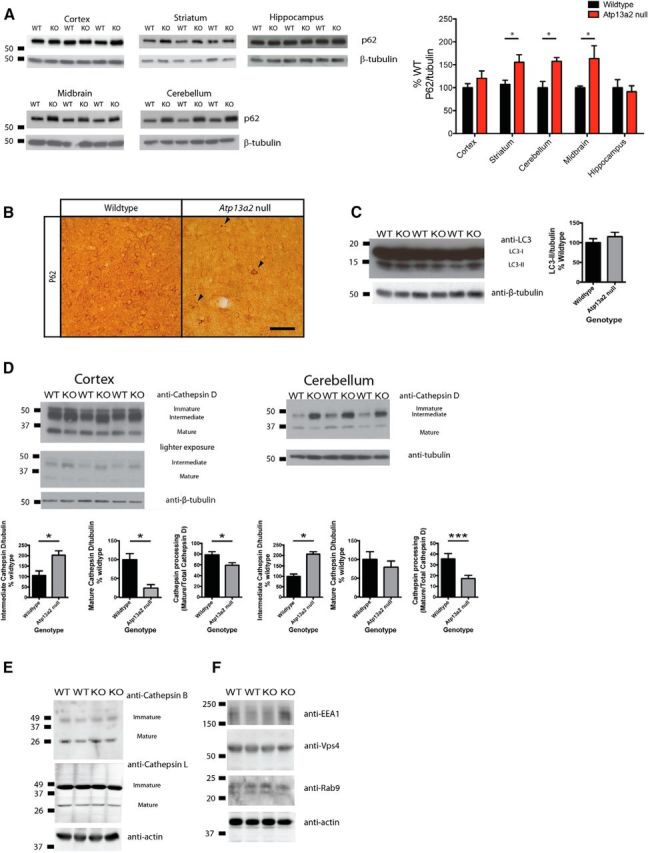
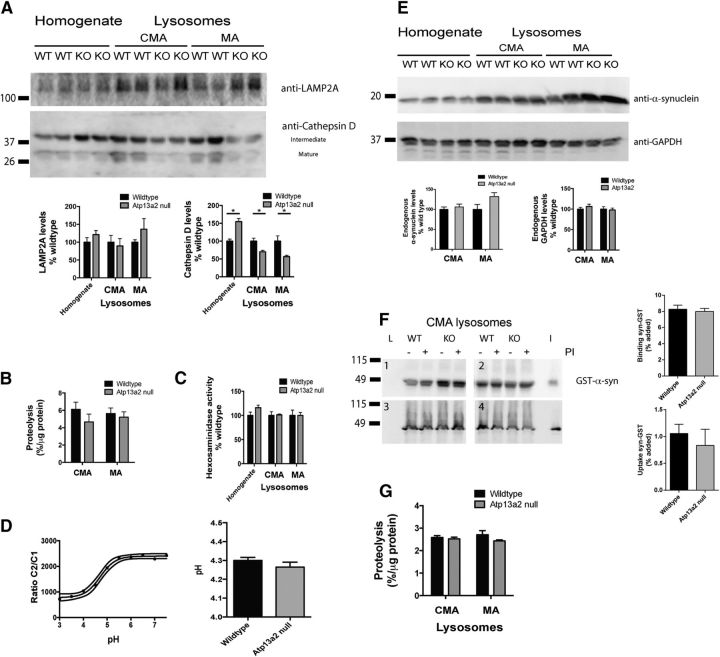
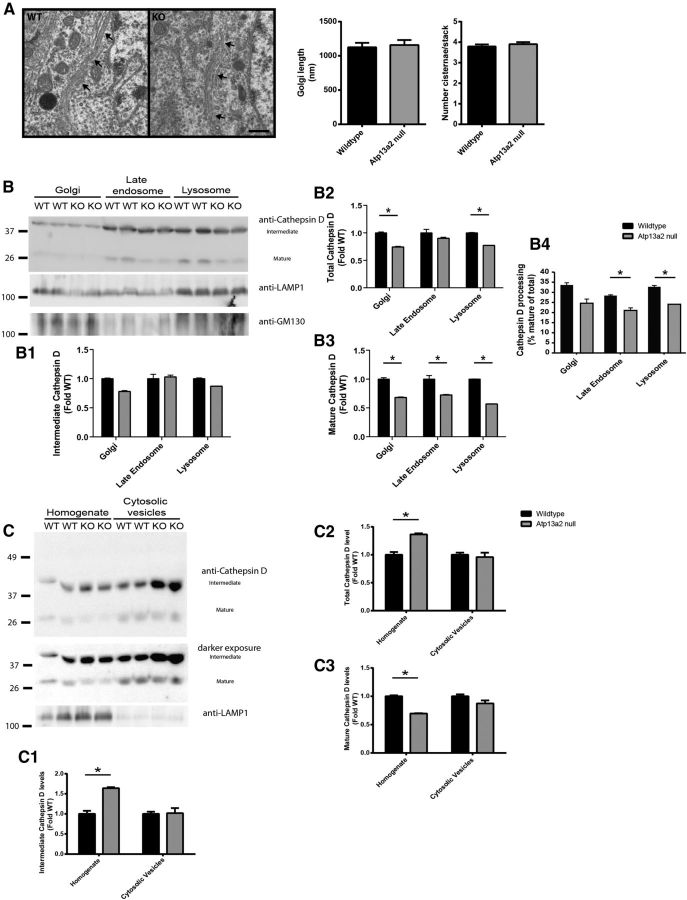
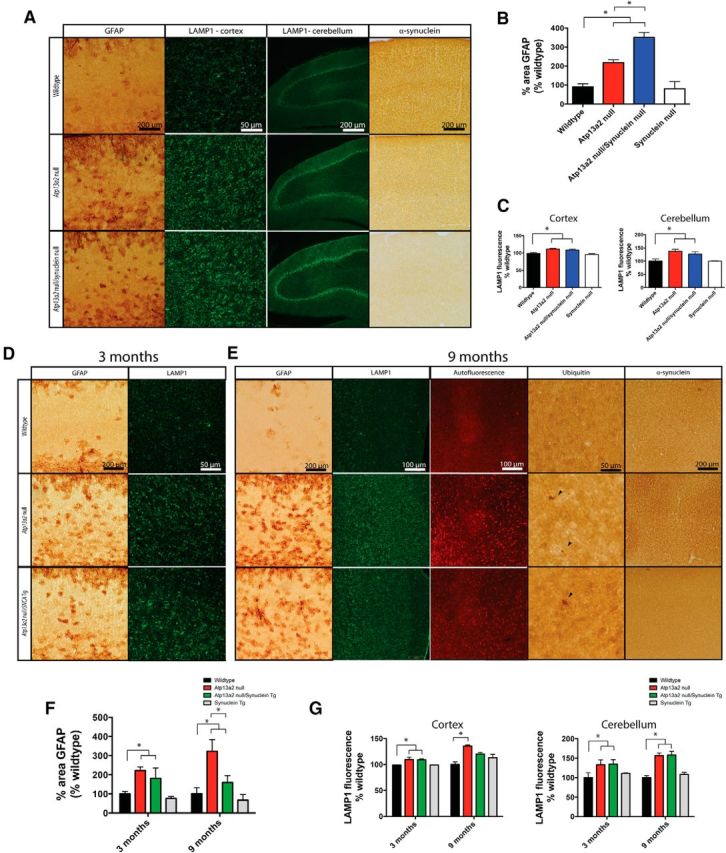
Accumulating evidence from genetic and biochemical studies implicates dysfunction of the autophagic-lysosomal pathway as a key feature in the pathogenesis of Parkinson's disease (PD). Most studies have focused on accumulation of neurotoxic α-synuclein secondary to defects in autophagy as the cause of neurodegeneration, but abnormalities of the autophagic-lysosomal system likely mediate toxicity through multiple mechanisms. To further explore how endolysosomal dysfunction causes PD-related neurodegeneration, we generated a murine model of Kufor-Rakeb syndrome (KRS), characterized by early-onset Parkinsonism with additional neurological features. KRS is caused by recessive loss-of-function mutations in the ATP13A2 gene encoding the endolysosomal ATPase ATP13A2. We show that loss of ATP13A2 causes a specific protein trafficking defect, and that Atp13a2 null mice develop age-related motor dysfunction that is preceded by neuropathological changes, including gliosis, accumulation of ubiquitinated protein aggregates, lipofuscinosis, and endolysosomal abnormalities. Contrary to predictions from in vitro data, in vivo mouse genetic studies demonstrate that these phenotypes are α-synuclein independent. Our findings indicate that endolysosomal dysfunction and abnormalities of α-synuclein homeostasis are not synonymous, even in the context of an endolysosomal genetic defect linked to Parkinsonism, and highlight the presence of α-synuclein-independent neurotoxicity consequent to endolysosomal dysfunction.
Keywords: ATP13A2; Parkinson's disease; autophagy; endolysosomal system; genetics.
Copyright © 2015 the authors 0270-6474/15/355724-19$15.00/0.
Figures
References
-
- Behrens MI, Brüggemann N, Chana P, Venegas P, Kägi M, Parrao T, Orellana P, Garrido C, Rojas CV, Hauke J, Hahnen E, González R, Seleme N, Fernández V, Schmidt A, Binkofski F, Kömpf D, Kubisch C, Hagenah J, Klein C, Ramirez A. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord. 2010;25:1929–1937. doi: 10.1002/mds.22996. - DOI - PubMed
-
- Brüggemann N, Hagenah J, Reetz K, Schmidt A, Kasten M, Buchmann I, Eckerle S, Bähre M, Münchau A, Djarmati A, van der Vegt J, Siebner H, Binkofski F, Ramirez A, Behrens MI, Klein C. Recessively inherited Parkinsonism: effect of ATP13A2 mutations on the clinical and neuroimaging phenotype. Arch Neurol. 2010;67:1357–1363. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials