

Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses
- PMID: 25856766
- PMCID: PMC4320458
- DOI: 10.1186/s12916-014-0259-2
Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses
Abstract
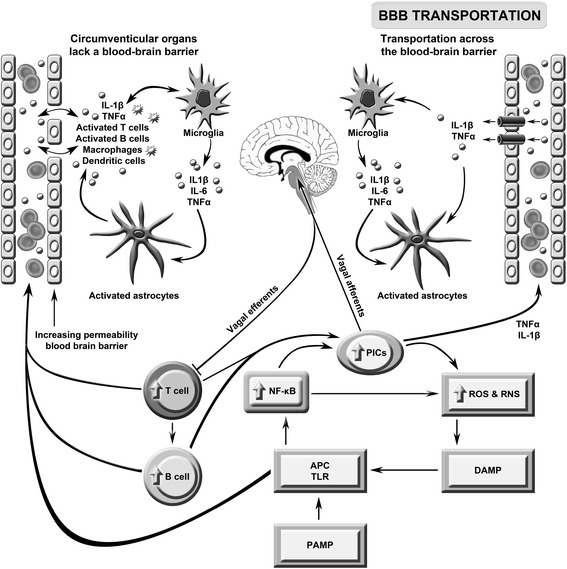
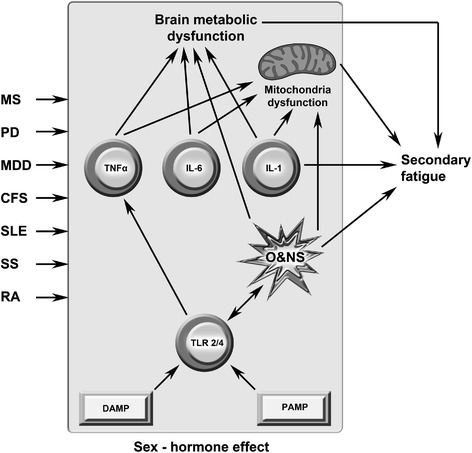
Background: The genesis of severe fatigue and disability in people following acute pathogen invasion involves the activation of Toll-like receptors followed by the upregulation of proinflammatory cytokines and the activation of microglia and astrocytes. Many patients suffering from neuroinflammatory and autoimmune diseases, such as multiple sclerosis, Parkinson's disease and systemic lupus erythematosus, also commonly suffer from severe disabling fatigue. Such patients also present with chronic peripheral immune activation and systemic inflammation in the guise of elevated proinflammtory cytokines, oxidative stress and activated Toll-like receptors. This is also true of many patients presenting with severe, apparently idiopathic, fatigue accompanied by profound levels of physical and cognitive disability often afforded the non-specific diagnosis of chronic fatigue syndrome.
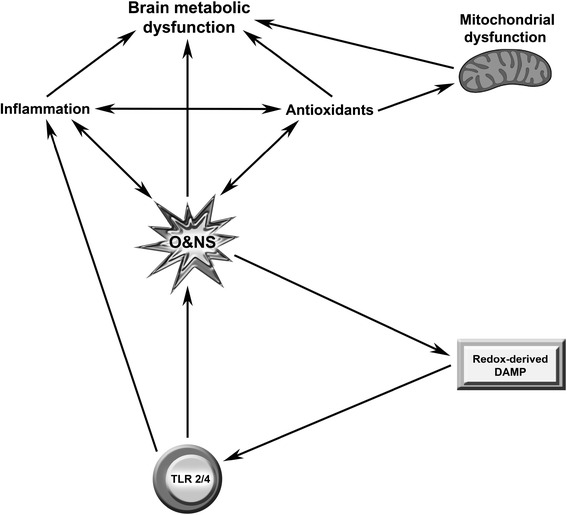
Discussion: Multiple lines of evidence demonstrate a positive association between the degree of peripheral immune activation, inflammation and oxidative stress, gray matter atrophy, glucose hypometabolism and cerebral hypoperfusion in illness, such as multiple sclerosis, Parkinson's disease and chronic fatigue syndrome. Most, if not all, of these abnormalities can be explained by a reduction in the numbers and function of astrocytes secondary to peripheral immune activation and inflammation. This is also true of the widespread mitochondrial dysfunction seen in otherwise normal tissue in neuroinflammatory, neurodegenerative and autoimmune diseases and in many patients with disabling, apparently idiopathic, fatigue. Given the strong association between peripheral immune activation and neuroinflammation with the genesis of fatigue the latter group of patients should be examined using FLAIR magnetic resonance imaging (MRI) and tested for the presence of peripheral immune activation.
Summary: It is concluded that peripheral inflammation and immune activation, together with the subsequent activation of glial cells and mitochondrial damage, likely account for the severe levels of intractable fatigue and disability seen in many patients with neuroimmune and autoimmune diseases.This would also appear to be the case for many patients afforded a diagnosis of Chronic Fatigue Syndrome.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
