

RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells
- PMID: 25858977
- PMCID: PMC4662681
- DOI: 10.1126/science.aaa6090
RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells
Abstract
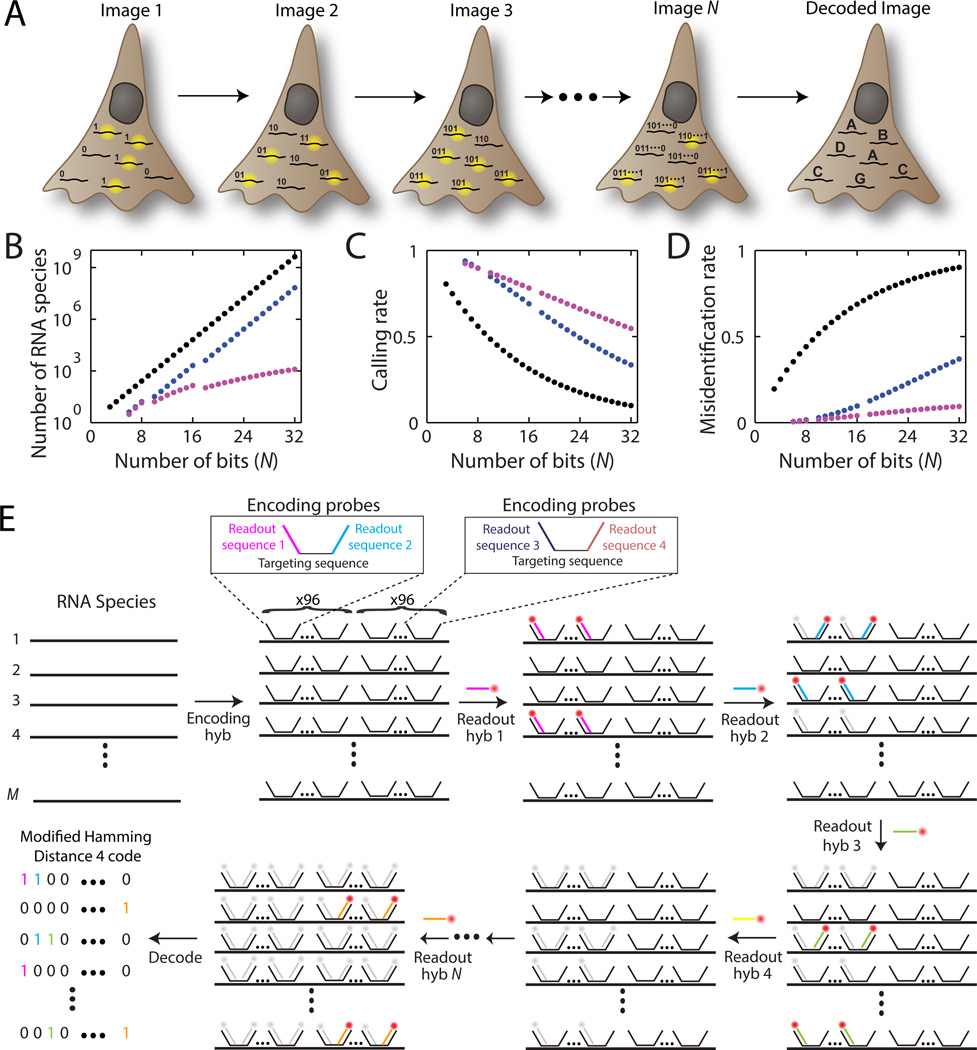
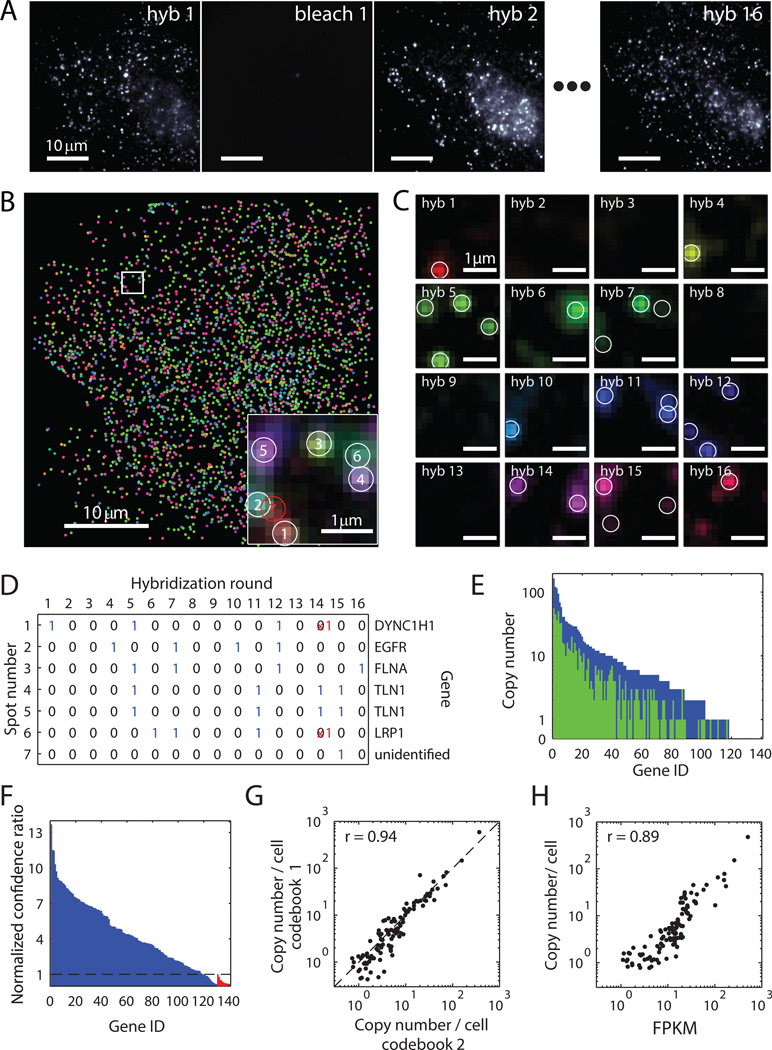
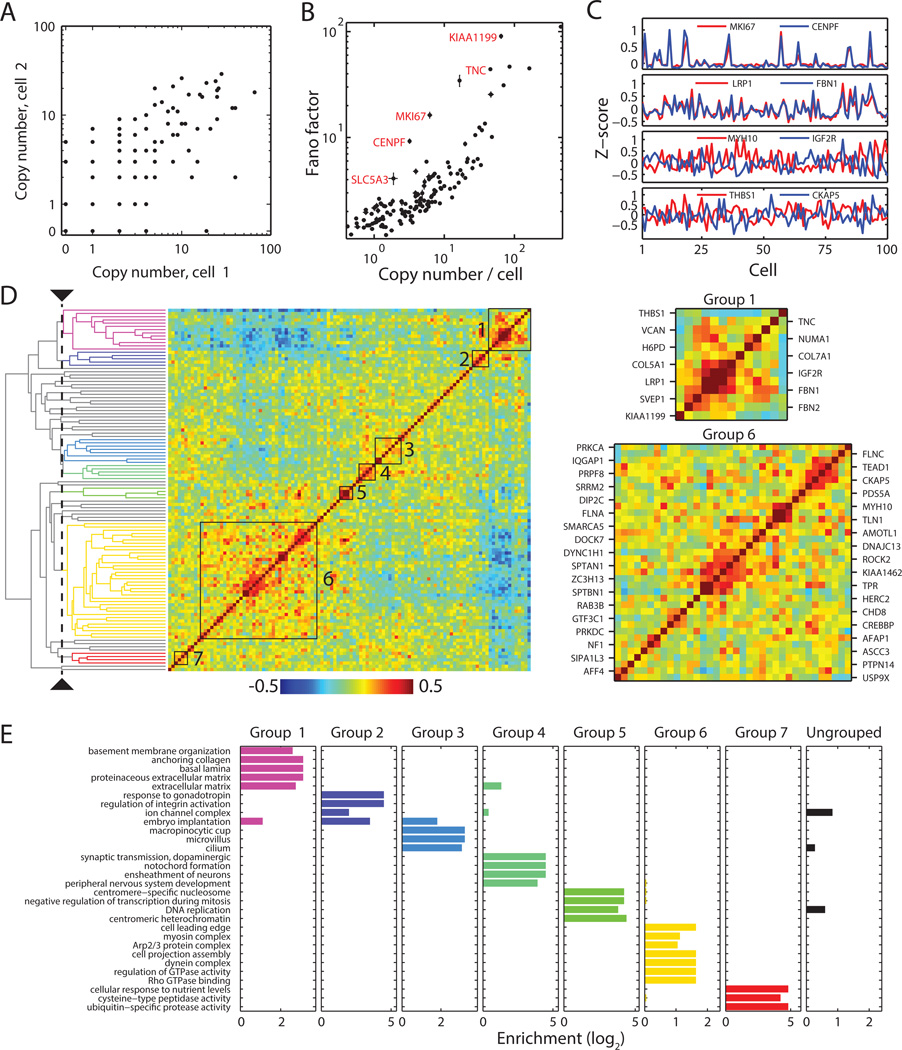
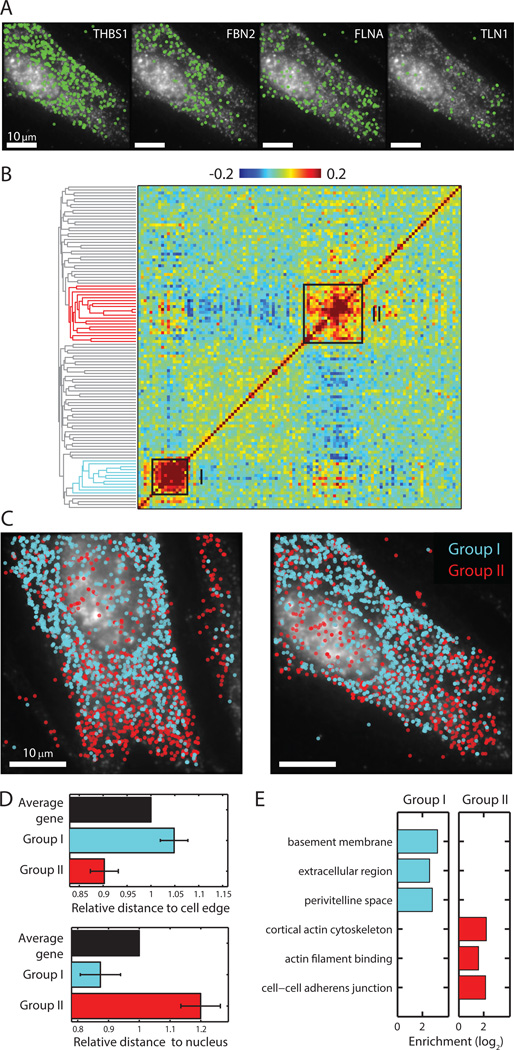
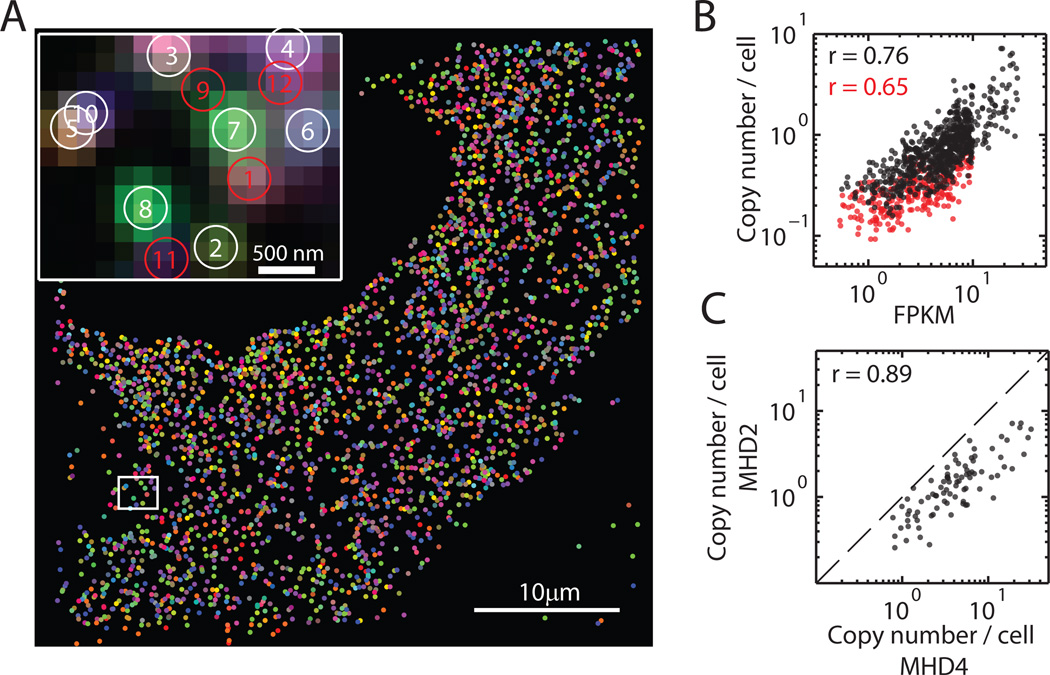
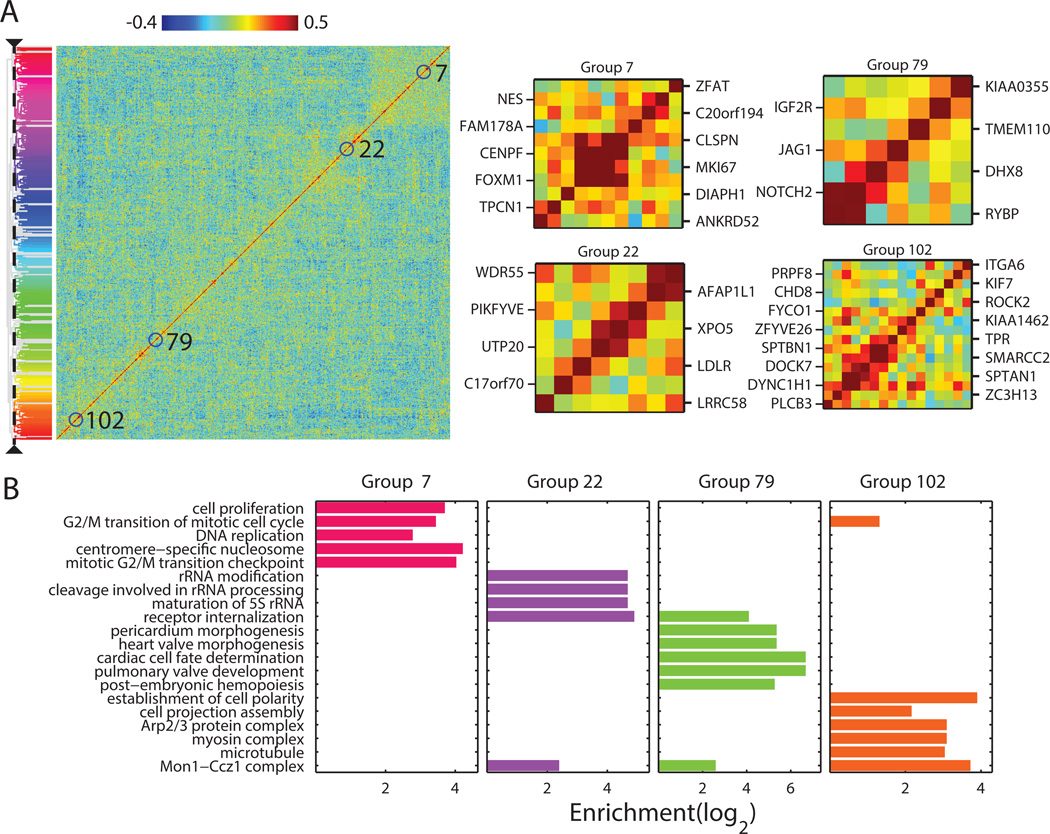
Knowledge of the expression profile and spatial landscape of the transcriptome in individual cells is essential for understanding the rich repertoire of cellular behaviors. Here, we report multiplexed error-robust fluorescence in situ hybridization (MERFISH), a single-molecule imaging approach that allows the copy numbers and spatial localizations of thousands of RNA species to be determined in single cells. Using error-robust encoding schemes to combat single-molecule labeling and detection errors, we demonstrated the imaging of 100 to 1000 distinct RNA species in hundreds of individual cells. Correlation analysis of the ~10(4) to 10(6) pairs of genes allowed us to constrain gene regulatory networks, predict novel functions for many unannotated genes, and identify distinct spatial distribution patterns of RNAs that correlate with properties of the encoded proteins.
Copyright © 2015, American Association for the Advancement of Science.
Figures
Comment in
-
RNA: Putting transcriptomics in its place.Nat Rev Genet. 2015 Jun;16(6):319. doi: 10.1038/nrg3951. Epub 2015 May 7. Nat Rev Genet. 2015. PMID: 25948245 No abstract available.
-
MERFISHing for spatial context.Trends Immunol. 2015 Jul;36(7):390-1. doi: 10.1016/j.it.2015.05.002. Epub 2015 May 23. Trends Immunol. 2015. PMID: 26013647
-
Highly multiplexed transcriptome imaging.Nat Methods. 2015 Jun;12(6):486-7. doi: 10.1038/nmeth.3426. Nat Methods. 2015. PMID: 26221655 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
