

Multiplex bisulfite PCR resequencing of clinical FFPE DNA
- PMID: 25861392
- PMCID: PMC4389706
- DOI: 10.1186/s13148-015-0067-3
Multiplex bisulfite PCR resequencing of clinical FFPE DNA
Abstract
Background: The clinical utility of DNA methylation as a predictive or prognostic biomarker requires scalable resequencing protocols for bisulfite-converted DNA. Key features of any validation method should be adaptability for low- or high-throughput needs and high reproducibility, and should only require minimal amounts of precious clinical sample as input material. Critically, this method should also deliver robust results when working with bisulfite-converted DNA extracted from formalin-fixed, paraffin-embedded (FFPE) blocks.
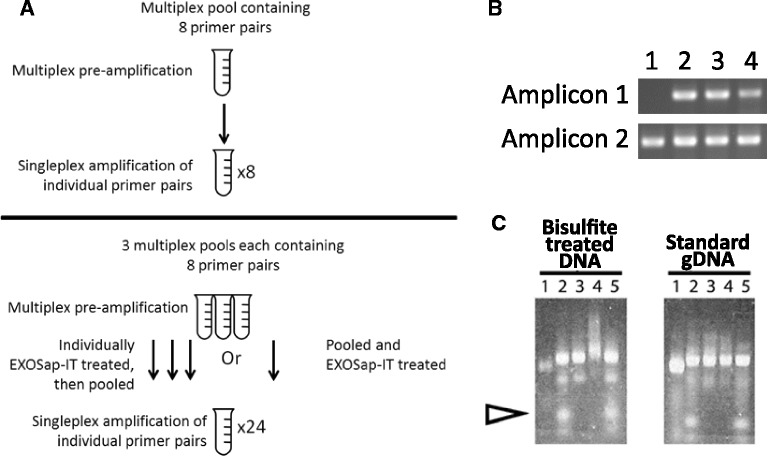
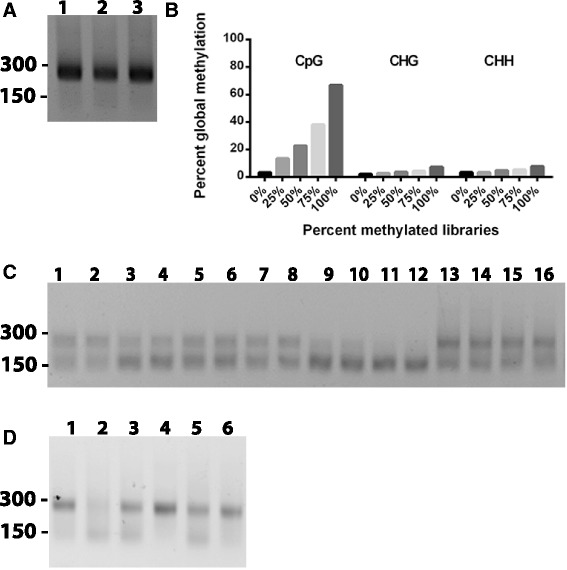
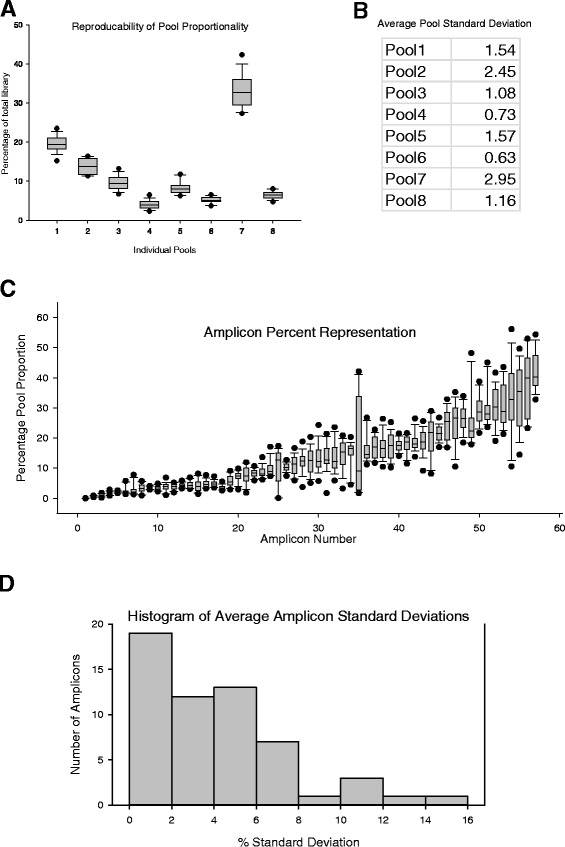
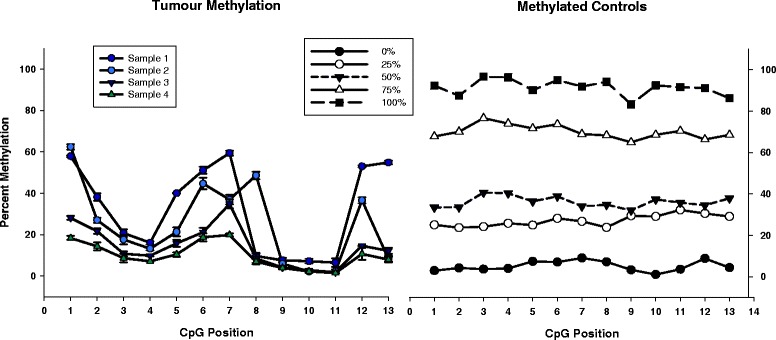
Results: We report here for the first time on comparison studies between the Fluidigm Access Array system and multiplex assays for multiplex bisulfite PCR resequencing. The requirement of the Fluidigm Access Array system for high template amounts and its sensitivity to variations in template quality rendered it unsuitable for bisulfite PCR applications utilizing FFPE DNA. In response to this limitation, we established a multiplex bisulfite PCR assay capable of delivering robust methylation data using minimal amounts of FFPE clinical DNA. To evaluate the parameters and reproducibility of this assay, 57 amplicons were used to prepare sequencing libraries in triplicate for 13 FFPE tumour samples, as well as a series of 5 methylated controls (0%, 25%, 50%, 75%, and 100%). Analysis of this data demonstrated that this multiplex assay had high reproducibility (mean standard deviation of 1.4% for methylation values), was low cost, required low sample input (50 ng of DNA or less), and could be scaled for both low- and high-throughput needs. Notably, ExoSAP-IT (exonuclease I) treatment to remove residual primers in bisulfite resequencing libraries appeared to degrade the library and generate a high-molecular weight smear which may impact on the degree of methylation assessed.
Conclusions: Multiplex bisulfite PCR assays represent a convenient and scalable method for validation and screening of methylated DNA regions from archival FFPE DNA. Moreover, the library construction process detailed here can be rapidly optimized and implemented with a minimal amount of work, can be performed using the standard equipment found in any molecular biology laboratory, and can be easily adapted for use on both genomic DNA and bisulfite DNA applications. However, in preparing bisulfite libraries for sequencing, the use of ExoSAP-IT is not recommended due to potential off-target nuclease effects which may impact downstream methylation analysis.
Keywords: Bisulfite PCR; DNA methylation; FFPE DNA; Fluidigm Access Array; Multiplex PCR.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
