

New approaches to establish genetic causality
- PMID: 25864169
- PMCID: PMC4560679
- DOI: 10.1016/j.tcm.2015.02.013
New approaches to establish genetic causality
Abstract
Cardiovascular medicine has evolved rapidly in the era of genomics with many diseases having primary genetic origins becoming the subject of intense investigation. The resulting avalanche of information on the molecular causes of these disorders has prompted a revolution in our understanding of disease mechanisms and provided new avenues for diagnoses. At the heart of this revolution is the need to correctly classify genetic variants discovered during the course of research or reported from clinical genetic testing. This review will address current concepts related to establishing the cause and effect relationship between genomic variants and heart diseases. A survey of general approaches used for functional annotation of variants will also be presented.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
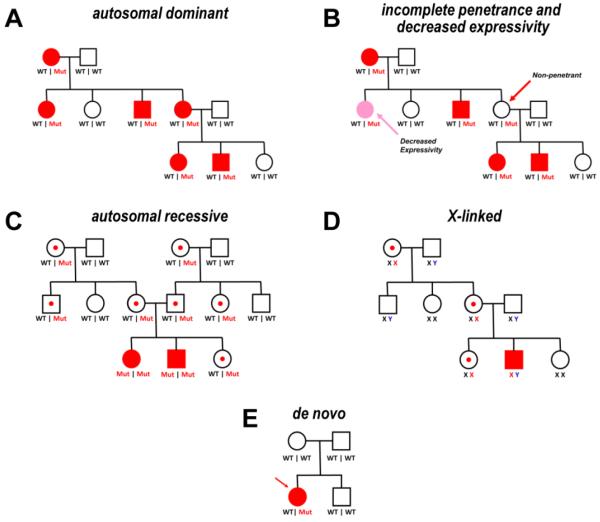

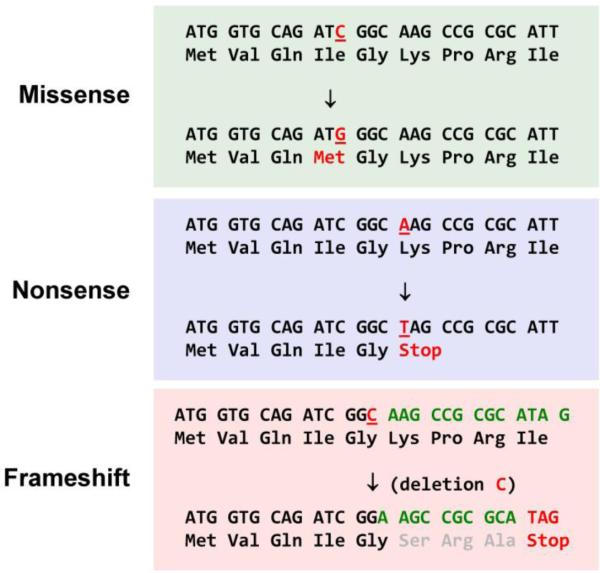
Comment in
-
Determining pathogenicity in cardiac genetic testing: Filling in the blank spaces.Trends Cardiovasc Med. 2015 Oct;25(7):653-4. doi: 10.1016/j.tcm.2015.03.004. Epub 2015 Mar 6. Trends Cardiovasc Med. 2015. PMID: 25850977 No abstract available.
Similar articles
-
Exploring predisposition and treatment response--the promise of genomics.Prog Cardiovasc Dis. 2012 Jul-Aug;55(1):56-63. doi: 10.1016/j.pcad.2012.04.006. Prog Cardiovasc Dis. 2012. PMID: 22824110 Free PMC article. Review.
-
Precision Medicine, Cardiovascular Disease and Hunting Elephants.Prog Cardiovasc Dis. 2016 May-Jun;58(6):651-60. doi: 10.1016/j.pcad.2016.02.004. Epub 2016 Feb 21. Prog Cardiovasc Dis. 2016. PMID: 26902518 Review.
-
Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults.Circ Cardiovasc Genet. 2017 Feb;10(1):e001573. doi: 10.1161/CIRCGENETICS.116.001573. Circ Cardiovasc Genet. 2017. PMID: 28087566 Free PMC article.
-
Cardiovascular genetic medicine: evolving concepts, rationale, and implementation.J Cardiovasc Transl Res. 2008 Jun;1(2):137-43. doi: 10.1007/s12265-008-9031-3. Epub 2008 May 20. J Cardiovasc Transl Res. 2008. PMID: 20559908 Review.
-
Genetics and Genomics of Single-Gene Cardiovascular Diseases: Common Hereditary Cardiomyopathies as Prototypes of Single-Gene Disorders.J Am Coll Cardiol. 2016 Dec 27;68(25):2831-2849. doi: 10.1016/j.jacc.2016.09.968. J Am Coll Cardiol. 2016. PMID: 28007145 Free PMC article. Review.
Cited by
-
Protein haploinsufficiency drivers identify MYBPC3 variants that cause hypertrophic cardiomyopathy.J Biol Chem. 2021 Jul;297(1):100854. doi: 10.1016/j.jbc.2021.100854. Epub 2021 Jun 5. J Biol Chem. 2021. PMID: 34097875 Free PMC article.
-
Genetic and phenotypic associations of frailty with cardiovascular indicators and behavioral characteristics.J Adv Res. 2025 May;71:263-277. doi: 10.1016/j.jare.2024.06.012. Epub 2024 Jun 9. J Adv Res. 2025. PMID: 38862035 Free PMC article.
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. - PubMed
-
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. - PubMed
-
- Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, McCormack L, et al. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15:972–977. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
