

Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state
- PMID: 25865119
- PMCID: PMC4403341
- DOI: 10.1038/ncomms7683
Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state
Abstract
Transcriptional reprogramming of proliferative melanoma cells into a phenotypically distinct invasive cell subpopulation is a critical event at the origin of metastatic spreading. Here we generate transcriptome, open chromatin and histone modification maps of melanoma cultures; and integrate this data with existing transcriptome and DNA methylation profiles from tumour biopsies to gain insight into the mechanisms underlying this key reprogramming event. This shows thousands of genomic regulatory regions underlying the proliferative and invasive states, identifying SOX10/MITF and AP-1/TEAD as regulators, respectively. Knockdown of TEADs shows a previously unrecognized role in the invasive gene network and establishes a causative link between these transcription factors, cell invasion and sensitivity to MAPK inhibitors. Using regulatory landscapes and in silico analysis, we show that transcriptional reprogramming underlies the distinct cellular states present in melanoma. Furthermore, it reveals an essential role for the TEADs, linking it to clinically relevant mechanisms such as invasion and resistance.
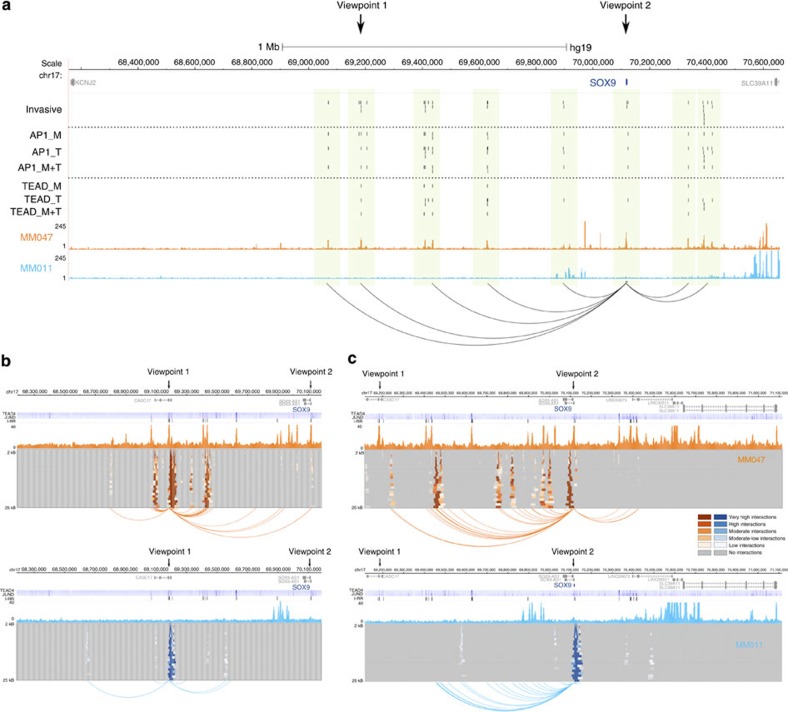
Figures







References
-
- Sun C. et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014) . - PubMed
-
- Widmer D. S. et al. Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 25, 343–353 (2012) . - PubMed
-
- Hoek K. S. et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68, 650–656 (2008) . - PubMed
-
- Caramel J. et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24, 466–480 (2013) . - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
