

Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains
- PMID: 25865888
- PMCID: PMC4408261
- DOI: 10.1016/j.celrep.2015.03.037
Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains
Abstract
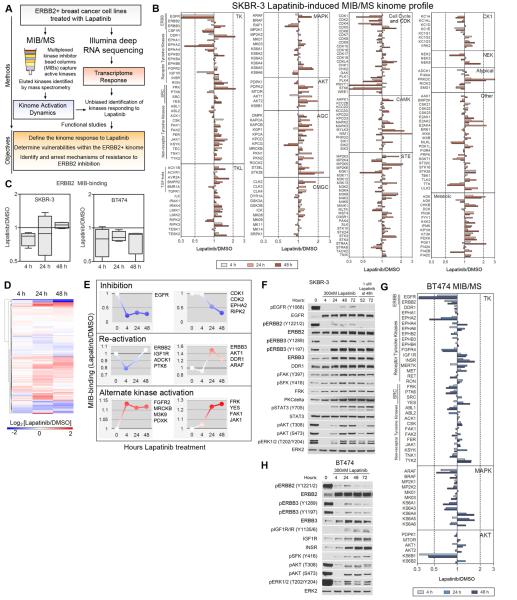
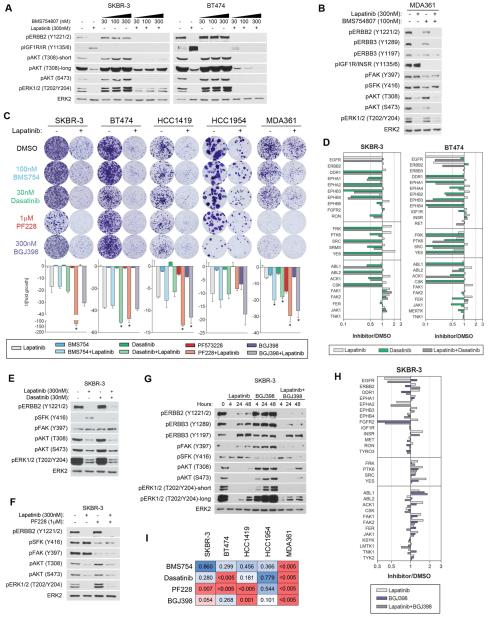
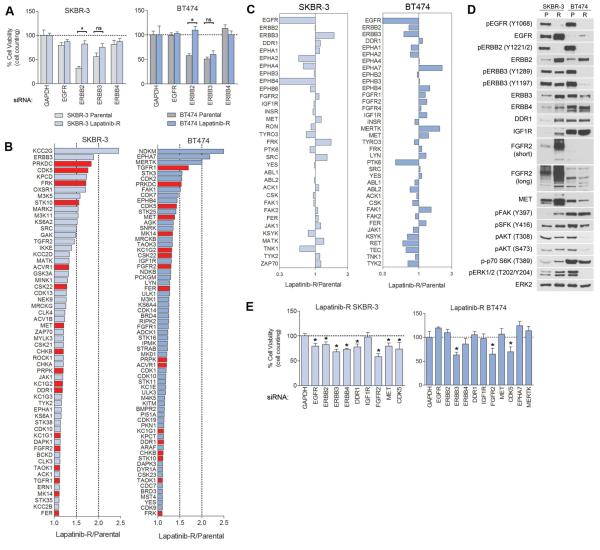
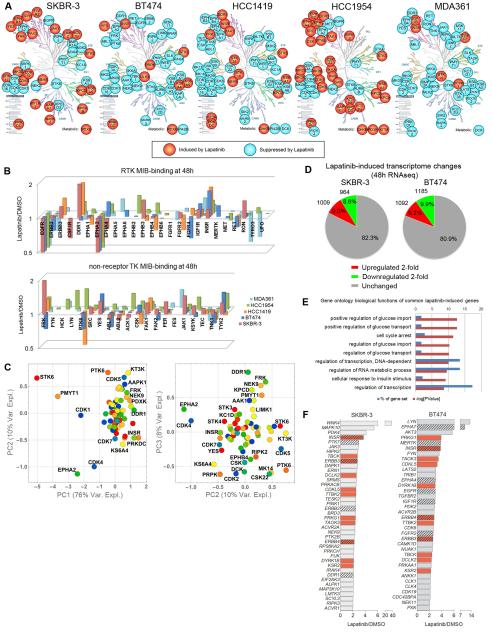
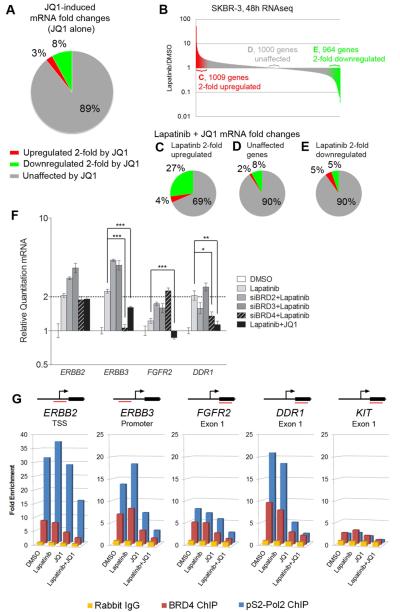
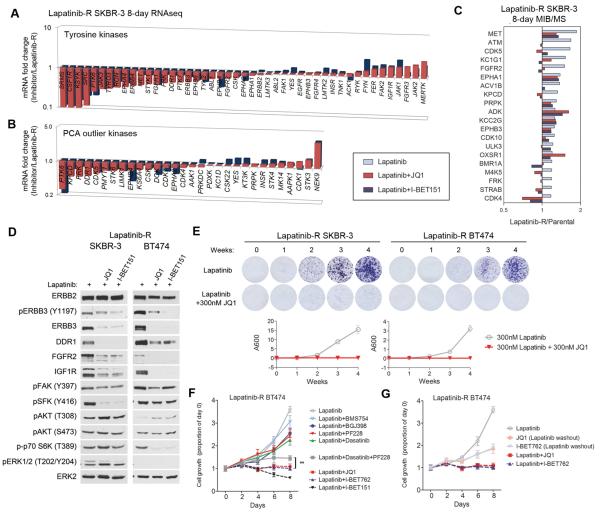
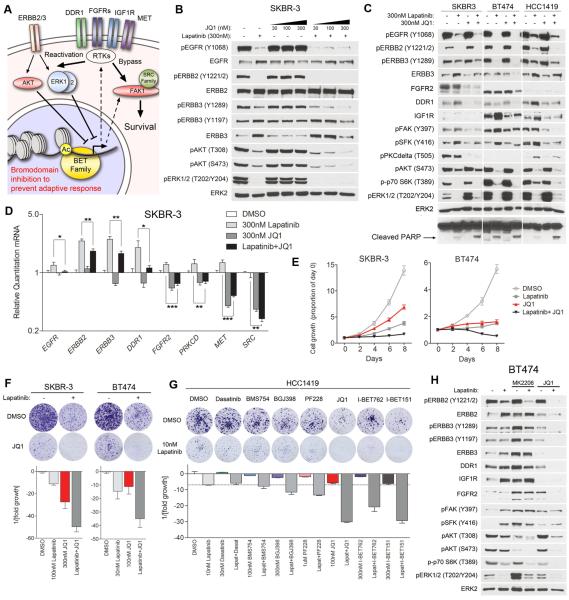
Therapeutics that target ERBB2, such as lapatinib, often provide initial clinical benefit, but resistance frequently develops. Adaptive responses leading to lapatinib resistance involve reprogramming of the kinome through reactivation of ERBB2/ERBB3 signaling and transcriptional upregulation and activation of multiple tyrosine kinases. The heterogeneity of induced kinases prevents their targeting by a single kinase inhibitor, underscoring the challenge of predicting effective kinase inhibitor combination therapies. We hypothesized that, to make the tumor response to single kinase inhibitors durable, the adaptive kinome response itself must be inhibited. Genetic and chemical inhibition of BET bromodomain chromatin readers suppresses transcription of many lapatinib-induced kinases involved in resistance, including ERBB3, IGF1R, DDR1, MET, and FGFRs, preventing downstream SRC/FAK signaling and AKT reactivation. Combining inhibitors of kinases and chromatin readers prevents kinome adaptation by blocking transcription, generating a durable response to lapatinib, and overcoming the dilemma of heterogeneity in the adaptive response.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Azuma K, Tsurutani J, Sakai K, Kaneda H, Fujisaka Y, Takeda M, Watatani M, Arao T, Satoh T, Okamoto I, et al. Switching addictions between HER2 and FGFR2 in HER2-positive breast tumor cells: FGFR2 as a potential target for salvage after lapatinib failure. Biochem Biophys Res Commun. 2011;407:219–224. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
