

Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development
- PMID: 25866365
- PMCID: PMC5019355
- DOI: 10.1016/j.pneurobio.2015.03.004
Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development
Abstract
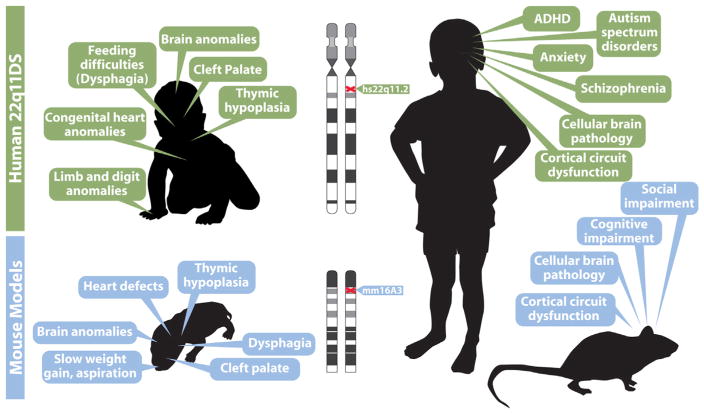
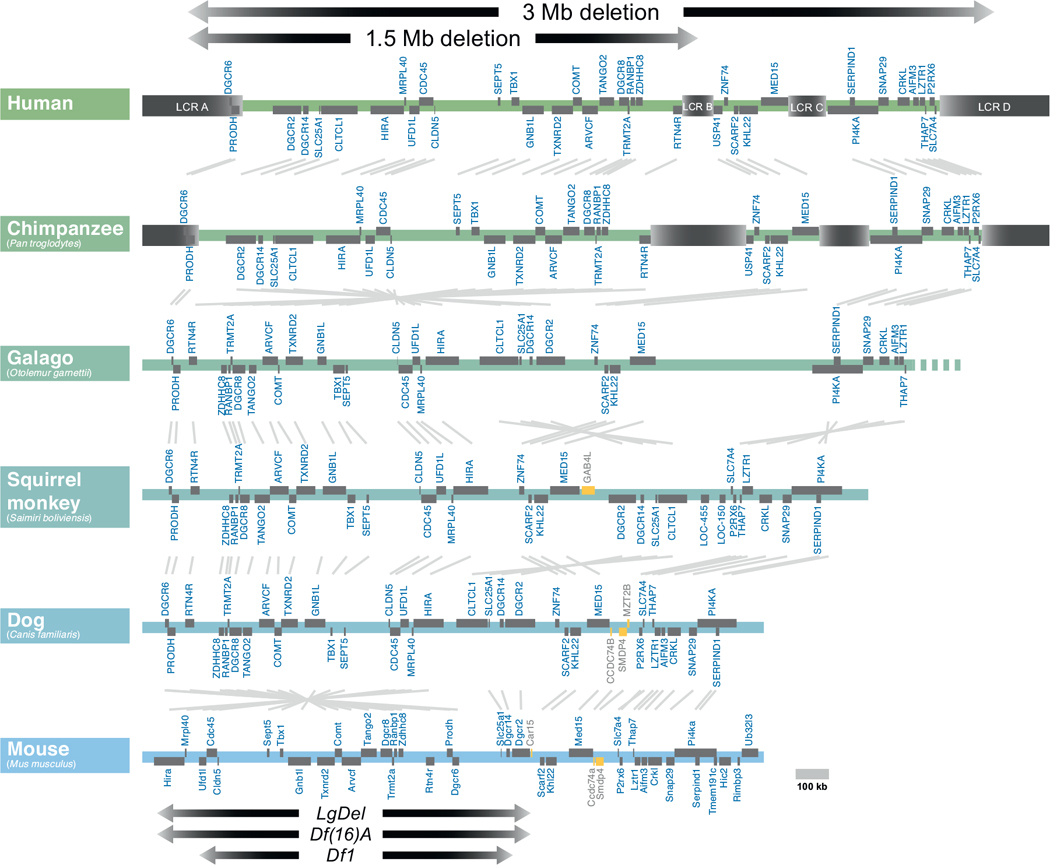
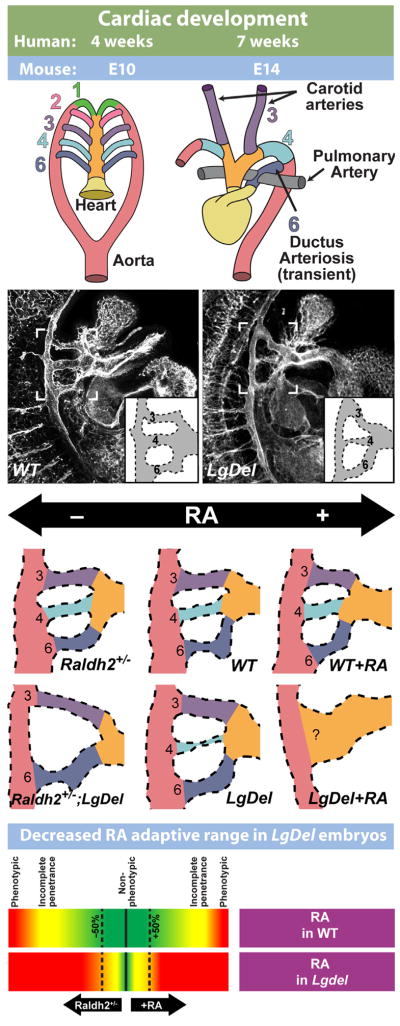
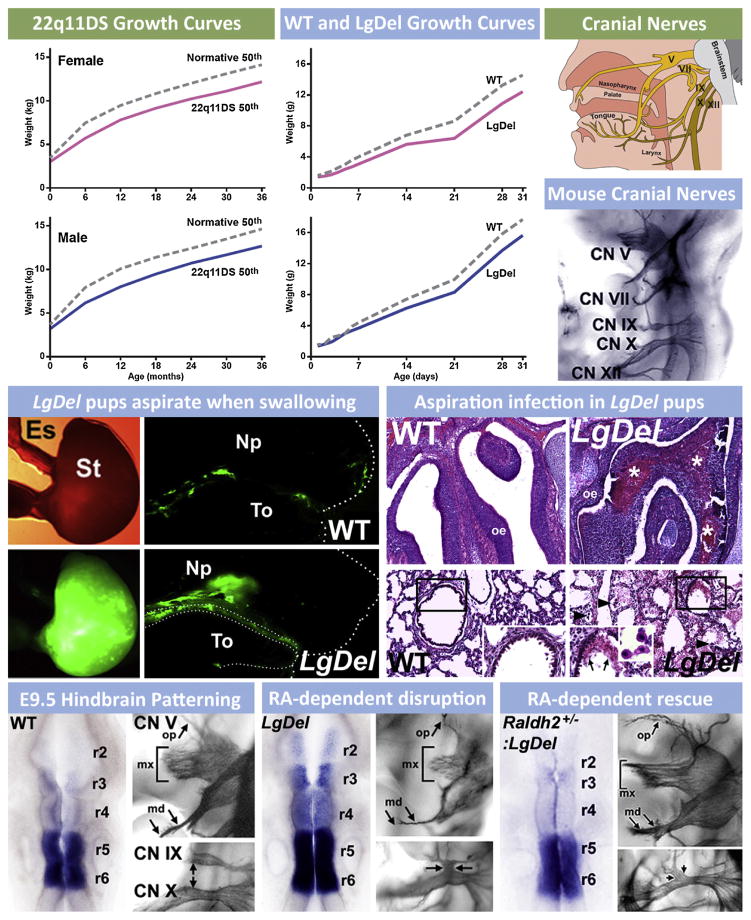
Understanding the developmental etiology of autistic spectrum disorders, attention deficit/hyperactivity disorder and schizophrenia remains a major challenge for establishing new diagnostic and therapeutic approaches to these common, difficult-to-treat diseases that compromise neural circuits in the cerebral cortex. One aspect of this challenge is the breadth and overlap of ASD, ADHD, and SCZ deficits; another is the complexity of mutations associated with each, and a third is the difficulty of analyzing disrupted development in at-risk or affected human fetuses. The identification of distinct genetic syndromes that include behavioral deficits similar to those in ASD, ADHC and SCZ provides a critical starting point for meeting this challenge. We summarize clinical and behavioral impairments in children and adults with one such genetic syndrome, the 22q11.2 Deletion Syndrome, routinely called 22q11DS, caused by micro-deletions of between 1.5 and 3.0 MB on human chromosome 22. Among many syndromic features, including cardiovascular and craniofacial anomalies, 22q11DS patients have a high incidence of brain structural, functional, and behavioral deficits that reflect cerebral cortical dysfunction and fall within the spectrum that defines ASD, ADHD, and SCZ. We show that developmental pathogenesis underlying this apparent genetic "model" syndrome in patients can be defined and analyzed mechanistically using genomically accurate mouse models of the deletion that causes 22q11DS. We conclude that "modeling a model", in this case 22q11DS as a model for idiopathic ASD, ADHD and SCZ, as well as other behavioral disorders like anxiety frequently seen in 22q11DS patients, in genetically engineered mice provides a foundation for understanding the causes and improving diagnosis and therapy for these disorders of cortical circuit development.
Keywords: ADHD; Animal models; Autism; Cortex; Schizophrenia.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
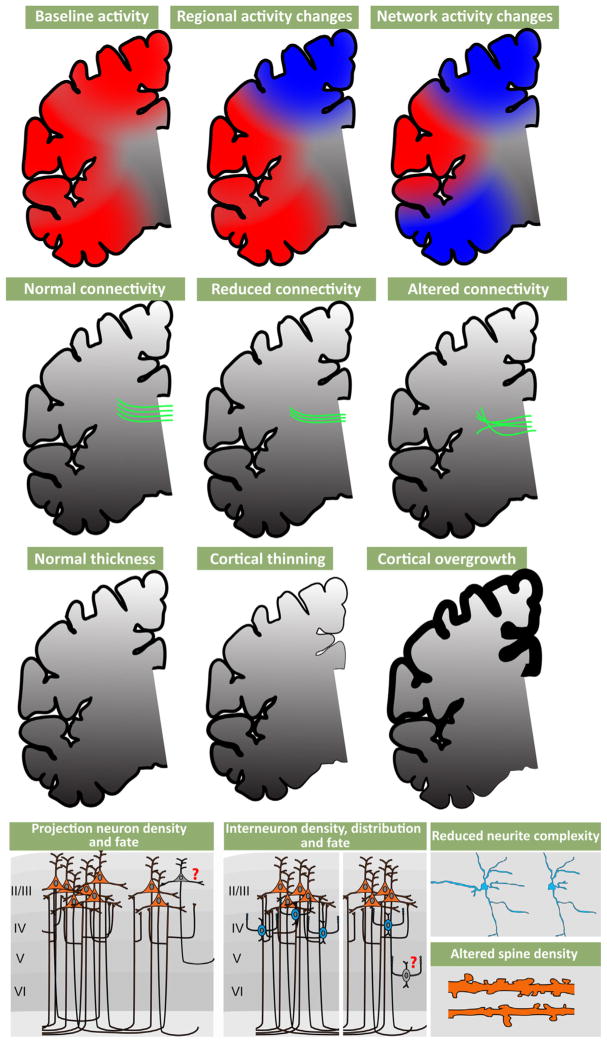
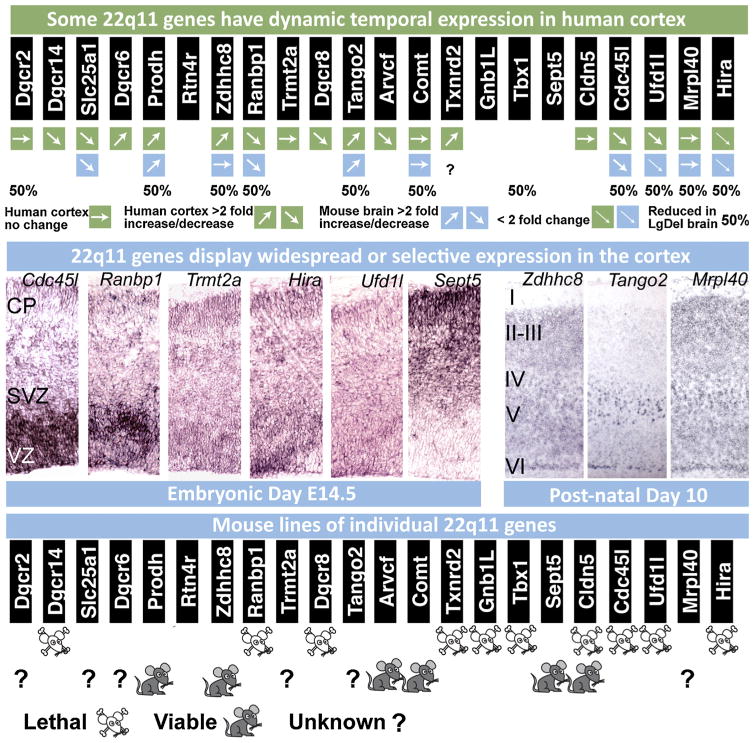
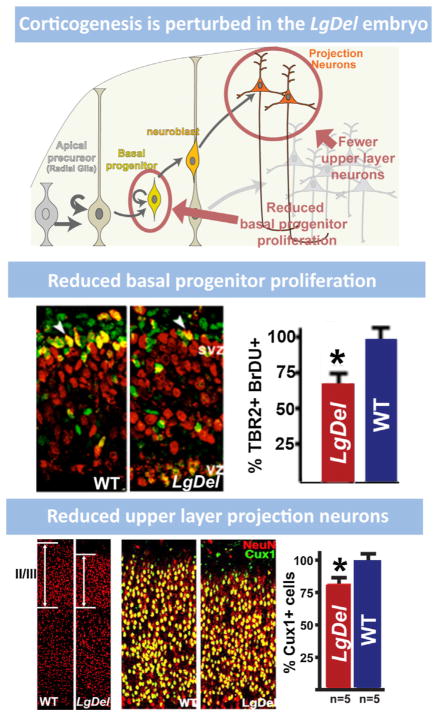
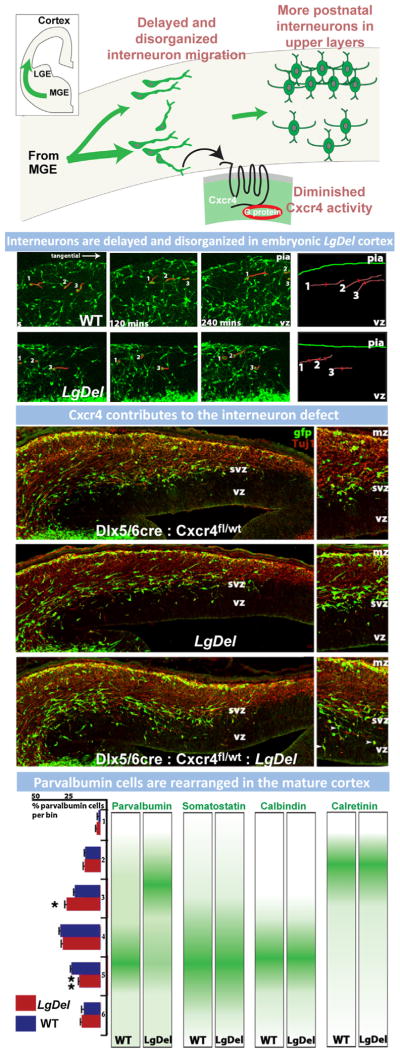
References
-
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Andersson F, Glaser B, Spiridon M, Debbane M, Vuilleumier P, Eliez S. Impaired activation of face processing networks revealed by functional magnetic resonance imaging in 22q11.2 deletion syndrome. Biol Psychiatry. 2008;63:49–57. - PubMed
-
- Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, Rujescu D, Werge T, van de Bunt M, Morris AP, McCarthy MI, Roddey JC, McEvoy LK, Desikan RS, Dale AM International Consortium for Blood Pressure G, Diabetes Genetics R, Meta-analysis C, Psychiatric Genomics Consortium Schizophrenia Working G. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. - PMC - PubMed
-
- Antshel KM, Aneja A, Strunge L, Peebles J, Fremont WP, Stallone K, Abdulsabur N, Higgins AM, Shprintzen RJ, Kates WR. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J Autism Dev Disord. 2007;37:1776–1786. - PubMed
Publication types
MeSH terms
Grants and funding
- HD029178/HD/NICHD NIH HHS/United States
- S10RR025565/RR/NCRR NIH HHS/United States
- P30 HD040677/HD/NICHD NIH HHS/United States
- R01 NS031768/NS/NINDS NIH HHS/United States
- HD042182/HD/NICHD NIH HHS/United States
- P30 NS045892/NS/NINDS NIH HHS/United States
- P50 MH064065/MH/NIMH NIH HHS/United States
- P30HD040677/HD/NICHD NIH HHS/United States
- S10 RR025565/RR/NCRR NIH HHS/United States
- MH64065/MH/NIMH NIH HHS/United States
- R01 HD029178/HD/NICHD NIH HHS/United States
- R29 HD029178/HD/NICHD NIH HHS/United States
- R01 DC011534/DC/NIDCD NIH HHS/United States
- P01 HD083157/HD/NICHD NIH HHS/United States
- NS031768/NS/NINDS NIH HHS/United States
- R01 HD042182/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
