

Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells
- PMID: 25866973
- PMCID: PMC4463197
- DOI: 10.1172/JCI78124
Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells
Abstract
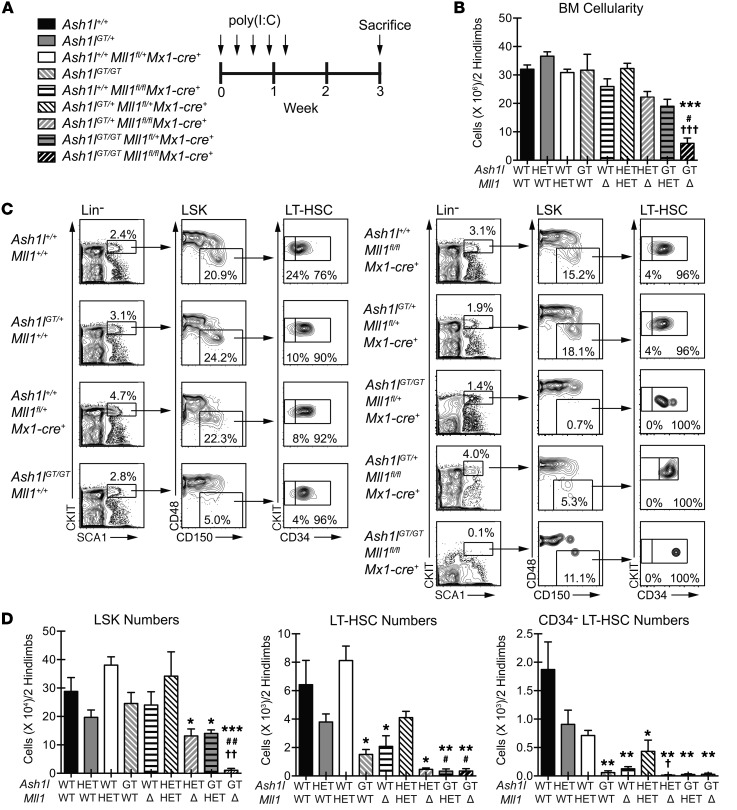
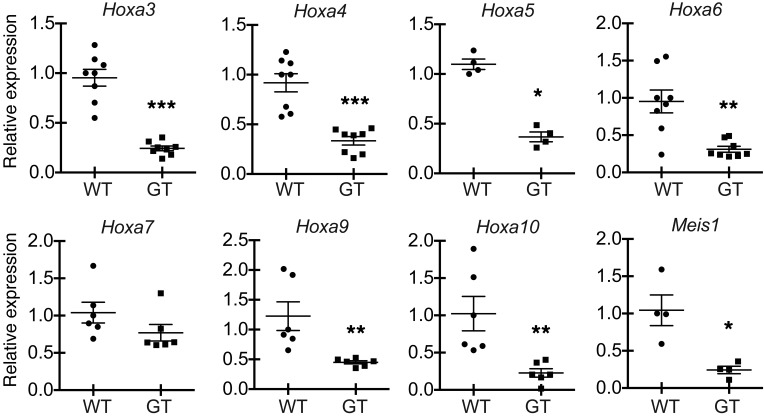
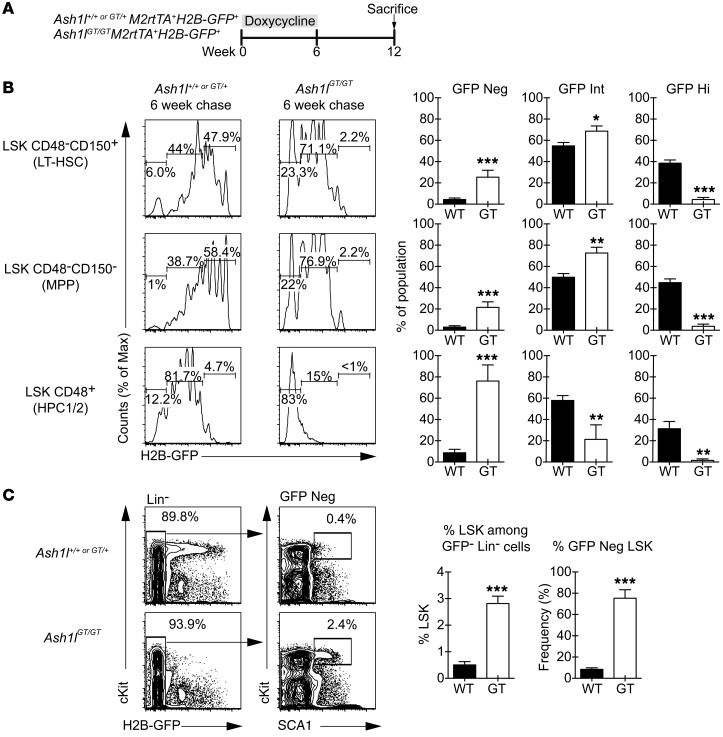
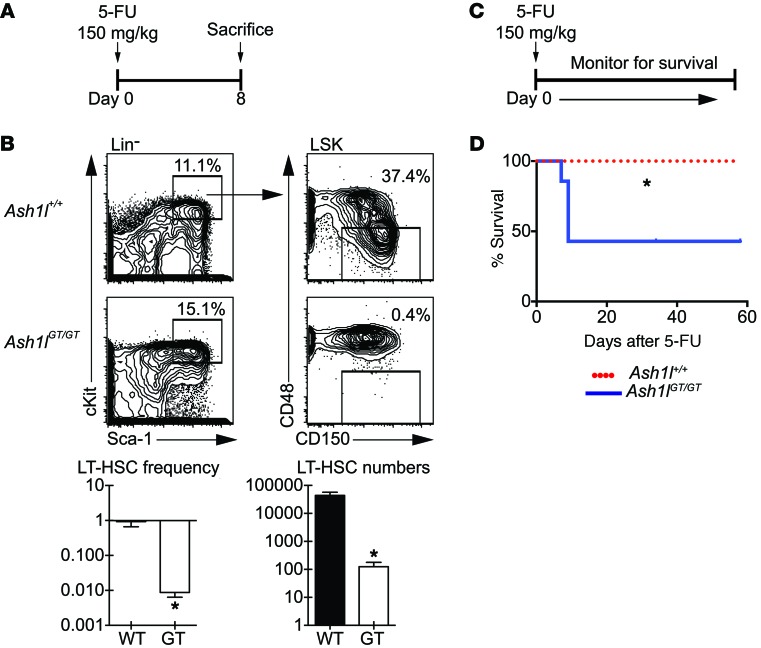
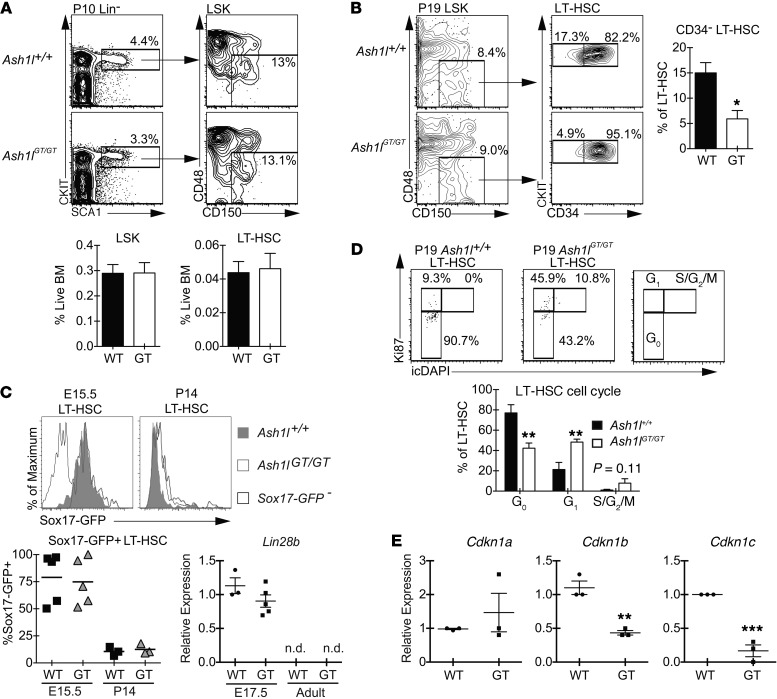
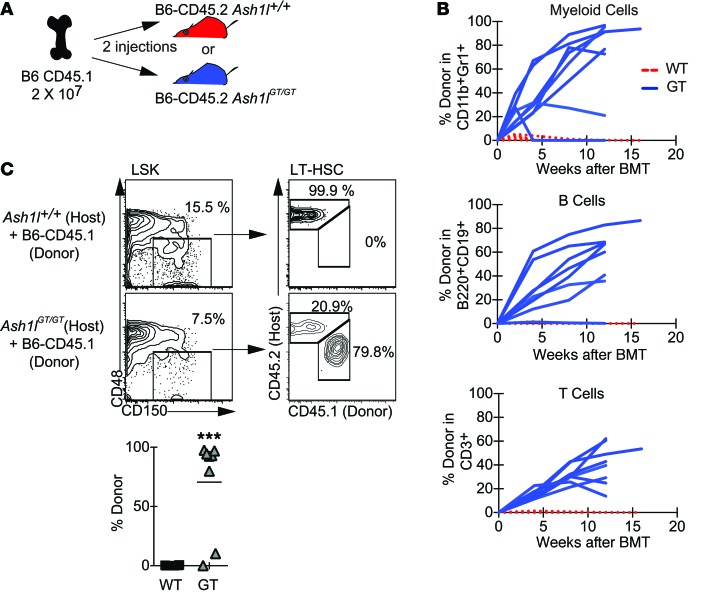
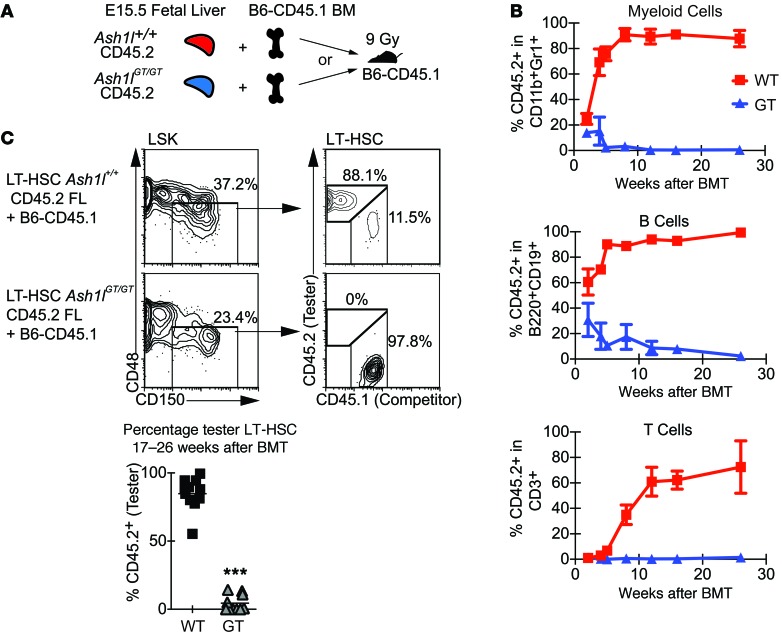
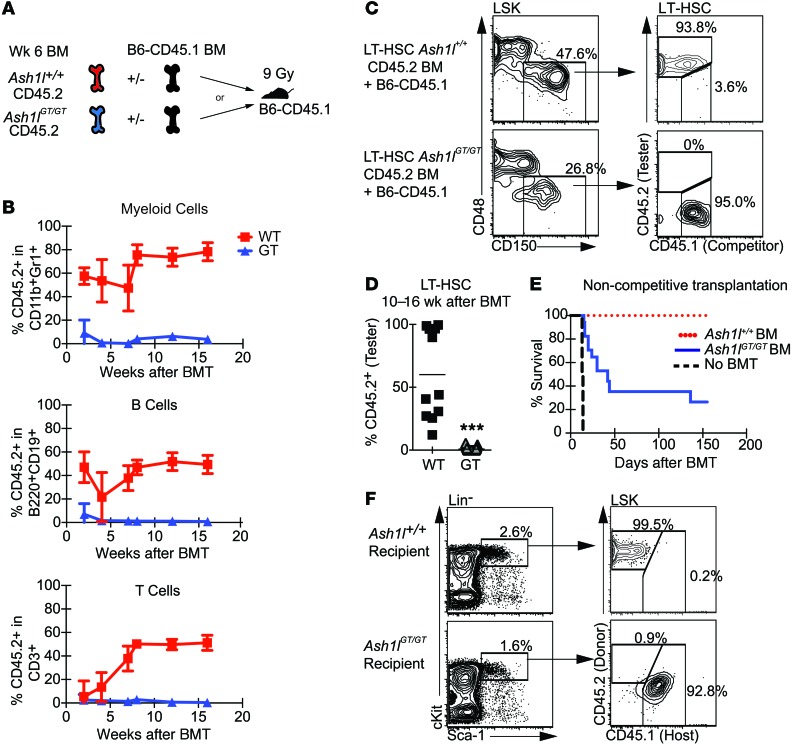
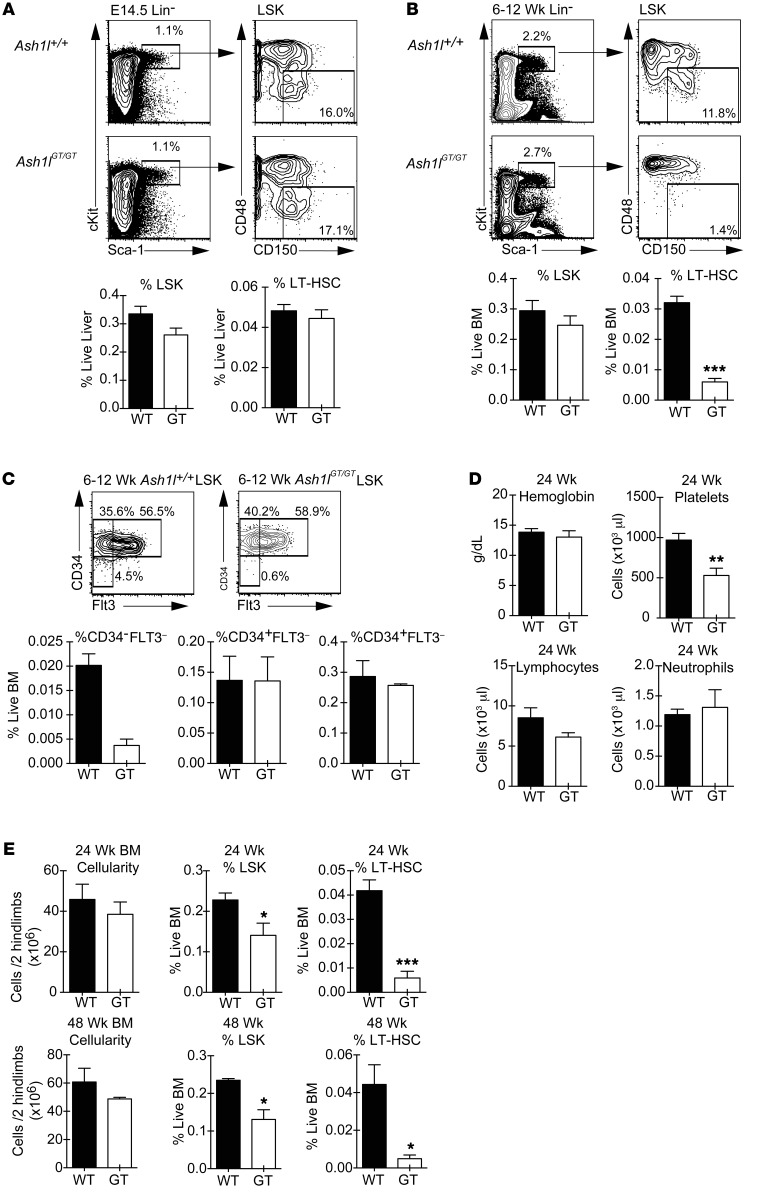
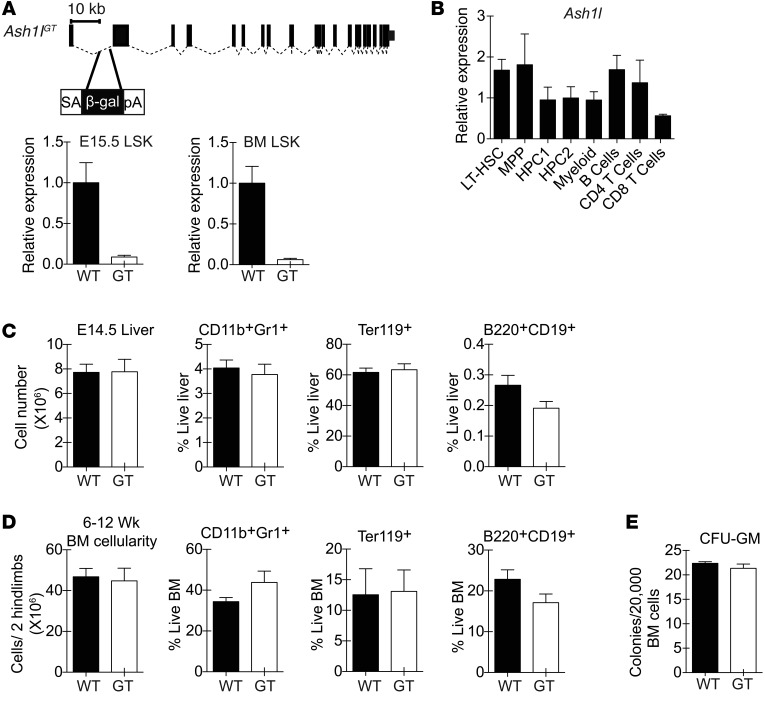
Rapidly cycling fetal and neonatal hematopoietic stem cells (HSCs) generate a pool of quiescent adult HSCs after establishing hematopoiesis in the bone marrow. We report an essential role for the trithorax group gene absent, small, or homeotic 1-like (Ash1l) at this developmental transition. Emergence and expansion of Ash1l-deficient fetal/neonatal HSCs were preserved; however, in young adult animals, HSCs were profoundly depleted. Ash1l-deficient adult HSCs had markedly decreased quiescence and reduced cyclin-dependent kinase inhibitor 1b/c (Cdkn1b/1c) expression and failed to establish long-term trilineage bone marrow hematopoiesis after transplantation to irradiated recipients. Wild-type HSCs could efficiently engraft when transferred to unirradiated, Ash1l-deficient recipients, indicating increased availability of functional HSC niches in these mice. Ash1l deficiency also decreased expression of multiple Hox genes in hematopoietic progenitors. Ash1l cooperated functionally with mixed-lineage leukemia 1 (Mll1), as combined loss of Ash1l and Mll1, but not isolated Ash1l or Mll1 deficiency, induced overt hematopoietic failure. Our results uncover a trithorax group gene network that controls quiescence, niche occupancy, and self-renewal potential in adult HSCs.
Figures
References
-
- Ema H, Nakauchi H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood. 2000;95(7):2284–2288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30-CA46592/CA/NCI NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- R01 GM082856/GM/NIGMS NIH HHS/United States
- R37-HD30428/HD/NICHD NIH HHS/United States
- R01 AI091627/AI/NIAID NIH HHS/United States
- R01 HD030428/HD/NICHD NIH HHS/United States
- GM07863/GM/NIGMS NIH HHS/United States
- T32 HD007505/HD/NICHD NIH HHS/United States
- T32 GM007863/GM/NIGMS NIH HHS/United States
- R37 HD030428/HD/NICHD NIH HHS/United States
- HD007505/HD/NICHD NIH HHS/United States
- GM007315/GM/NIGMS NIH HHS/United States
- R01-AI091627/AI/NIAID NIH HHS/United States
- T32 GM007315/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
