

Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations
- PMID: 25868664
- PMCID: PMC4841446
- DOI: 10.1007/s10545-015-9836-6
Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations
Abstract
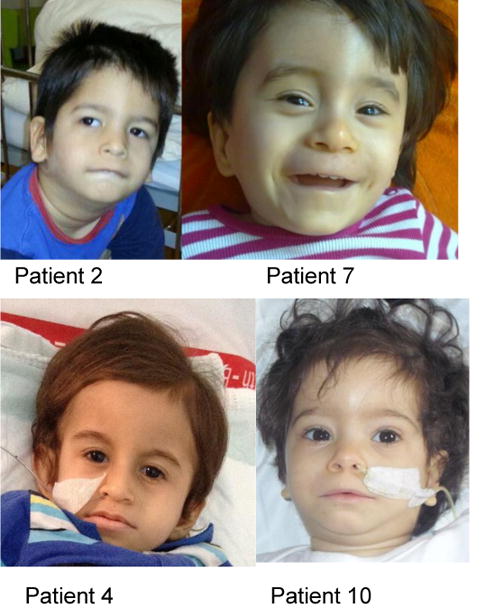
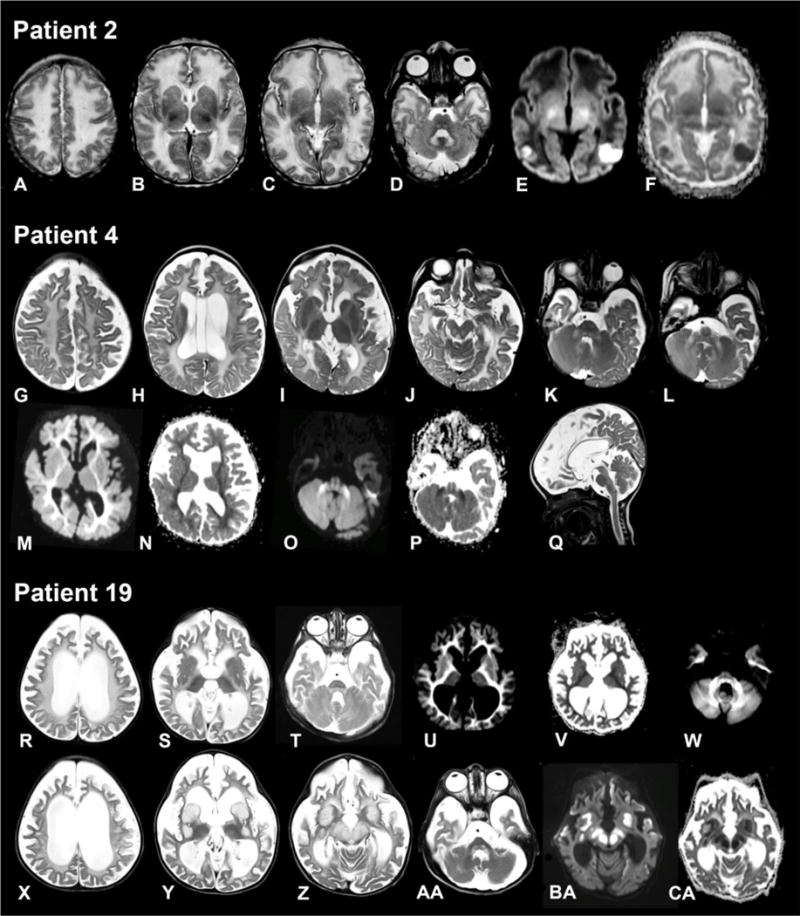
FBXL4 deficiency is a recently described disorder of mitochondrial maintenance associated with a loss of mitochondrial DNA in cells. To date, the genetic diagnosis of FBXL4 deficiency has been established in 28 individuals. This paper retrospectively reviews proxy-reported clinical and biochemical findings and evaluates brain imaging, morphological and genetic data in 21 of those patients. Neonatal/early-onset severe lactic acidosis, muscular hypotonia, feeding problems and failure to thrive is the characteristic pattern at first presentation. Facial dysmorphic features are present in 67% of cases. Seven children died (mean age 37 months); 11 children were alive (mean age at follow-up 46 months), three children were lost to follow-up. All survivors developed severe psychomotor retardation. Brain imaging was non-specific in neonates but a later-onset, rapidly progressive brain atrophy was noted. Elevated blood lactate and metabolic acidosis were observed in all individuals; creatine kinase was elevated in 45% of measurements. Diagnostic workup in patient tissues and cells revealed a severe combined respiratory chain defect with a general decrease of enzymes associated with mitochondrial energy metabolism and a relative depletion of mitochondrial DNA content. Mutations were detected throughout the FBXL4 gene albeit with no clear delineation of a genotype-phenotype correlation. Treatment with "mitochondrial medications" did not prove effective. In conclusion, a clinical pattern of early-onset encephalopathy, persistent lactic acidosis, profound muscular hypotonia and typical facial dysmorphism should prompt initiation of molecular genetic analysis of FBXL4. Establishment of the diagnosis permits genetic counselling, prevents patients undergoing unhelpful diagnostic procedures and allows for accurate prognosis.
Conflict of interest statement
Compliance with ethics guidelines Conflict of interest None.
Figures


References
-
- Acham-Roschitz B, Plecko B, Lindbichler F, Bittner R, Mache CJ, Sperl W, Mayr JA. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–304. - PubMed
-
- Barbier A, Boivin A, Yoon W, et al. New reference curves for head circumference at birth, by gestational age. New reference curves for head circumference at birth, by gestational age. Pediatrics. 2013;131:1158–1167. - PubMed
-
- Berger A, Mayr JA, Meierhofer D, et al. Severe depletion of mitochondrial DNA in spinal muscular atrophy. Acta Neuropathol. 2003;105:245–251. - PubMed
-
- Blau N, Duran M, Blaskovic ME, Gibson KM, editors. Physician’s guide to the laboratory diagnosis of metabolic diseases. 2nd. Springer; Berlin: 2003.
-
- Böhm M, Pronicka E, Karcmarewicz E, et al. Retrospective, multicentric study of 180 children with cytochrome c oxidase deficiency. Pediatr Res. 2006;59:21–26. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
