

Therapeutic targeting of replicative immortality
- PMID: 25869441
- PMCID: PMC4600408
- DOI: 10.1016/j.semcancer.2015.03.007
Therapeutic targeting of replicative immortality
Abstract
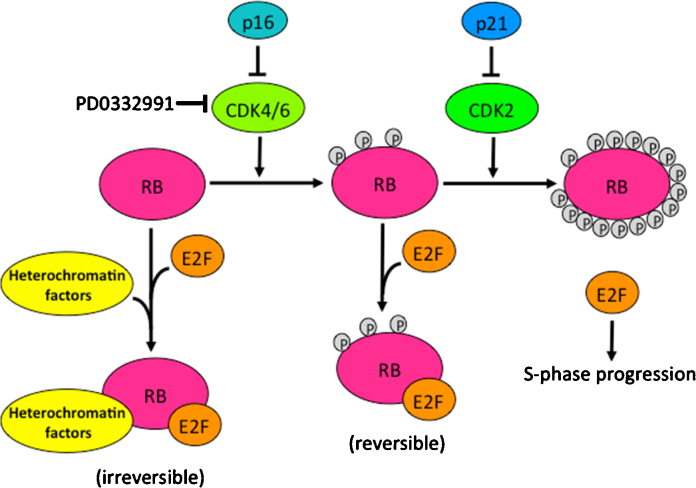
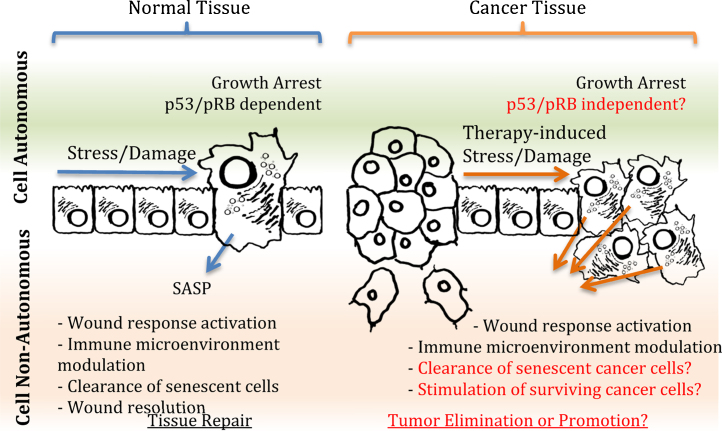
One of the hallmarks of malignant cell populations is the ability to undergo continuous proliferation. This property allows clonal lineages to acquire sequential aberrations that can fuel increasingly autonomous growth, invasiveness, and therapeutic resistance. Innate cellular mechanisms have evolved to regulate replicative potential as a hedge against malignant progression. When activated in the absence of normal terminal differentiation cues, these mechanisms can result in a state of persistent cytostasis. This state, termed "senescence," can be triggered by intrinsic cellular processes such as telomere dysfunction and oncogene expression, and by exogenous factors such as DNA damaging agents or oxidative environments. Despite differences in upstream signaling, senescence often involves convergent interdependent activation of tumor suppressors p53 and p16/pRB, but can be induced, albeit with reduced sensitivity, when these suppressors are compromised. Doses of conventional genotoxic drugs required to achieve cancer cell senescence are often much lower than doses required to achieve outright cell death. Additional therapies, such as those targeting cyclin dependent kinases or components of the PI3K signaling pathway, may induce senescence specifically in cancer cells by circumventing defects in tumor suppressor pathways or exploiting cancer cells' heightened requirements for telomerase. Such treatments sufficient to induce cancer cell senescence could provide increased patient survival with fewer and less severe side effects than conventional cytotoxic regimens. This positive aspect is countered by important caveats regarding senescence reversibility, genomic instability, and paracrine effects that may increase heterogeneity and adaptive resistance of surviving cancer cells. Nevertheless, agents that effectively disrupt replicative immortality will likely be valuable components of new combinatorial approaches to cancer therapy.
Keywords: Oncogenic stress; Senescence; Telomerase; p53; pRB.
Copyright © 2015 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures

References
-
- Wright W.E., Shay J.W. Time, telomeres and tumors: is cellular senescence more than an anticancer mechanism. Trends Cell Biol. 1995;5:293–297. - PubMed
-
- Kipling D., Wynford-Thomas D., Jones C.J., Akbar A., Aspinall R., Bacchetti S. Telomere-dependent senescence. Nat Biotechnol. 1999;17:313–314. - PubMed
-
- Morin G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. - PubMed
-
- Lingner J., Hughes T.R., Shevchenko A., Mann M., Lundblad V., Cech T.R. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
