

Excision of translesion synthesis errors orchestrates responses to helix-distorting DNA lesions
- PMID: 25869665
- PMCID: PMC4395481
- DOI: 10.1083/jcb.201408017
Excision of translesion synthesis errors orchestrates responses to helix-distorting DNA lesions
Abstract
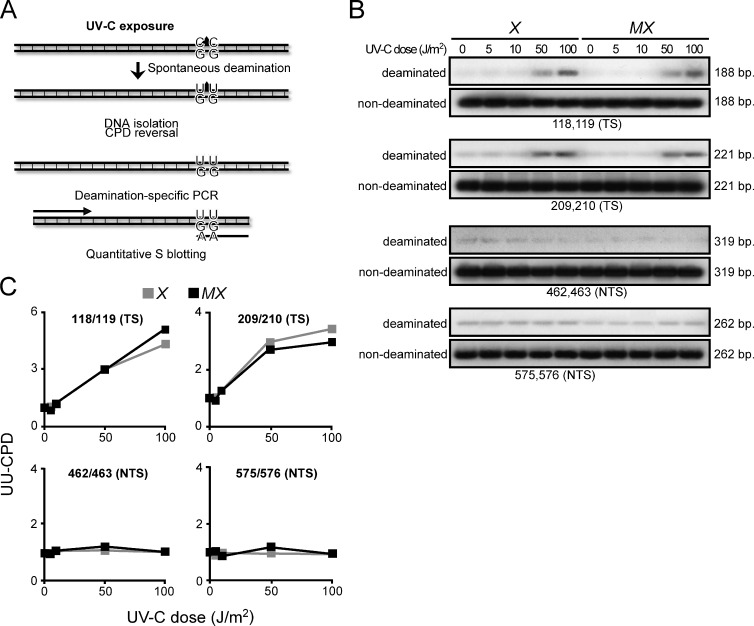
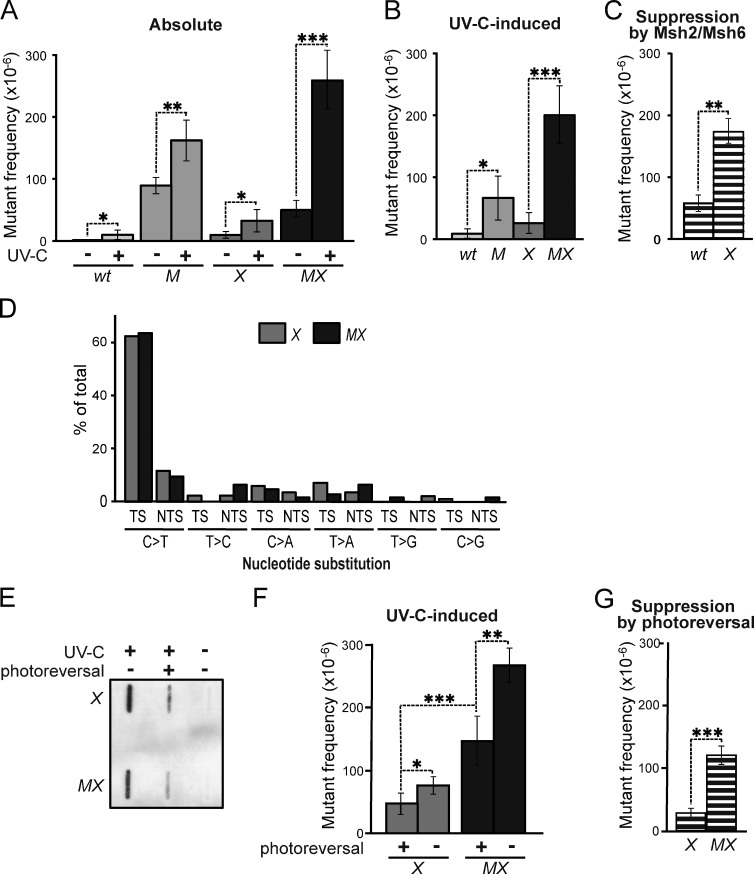
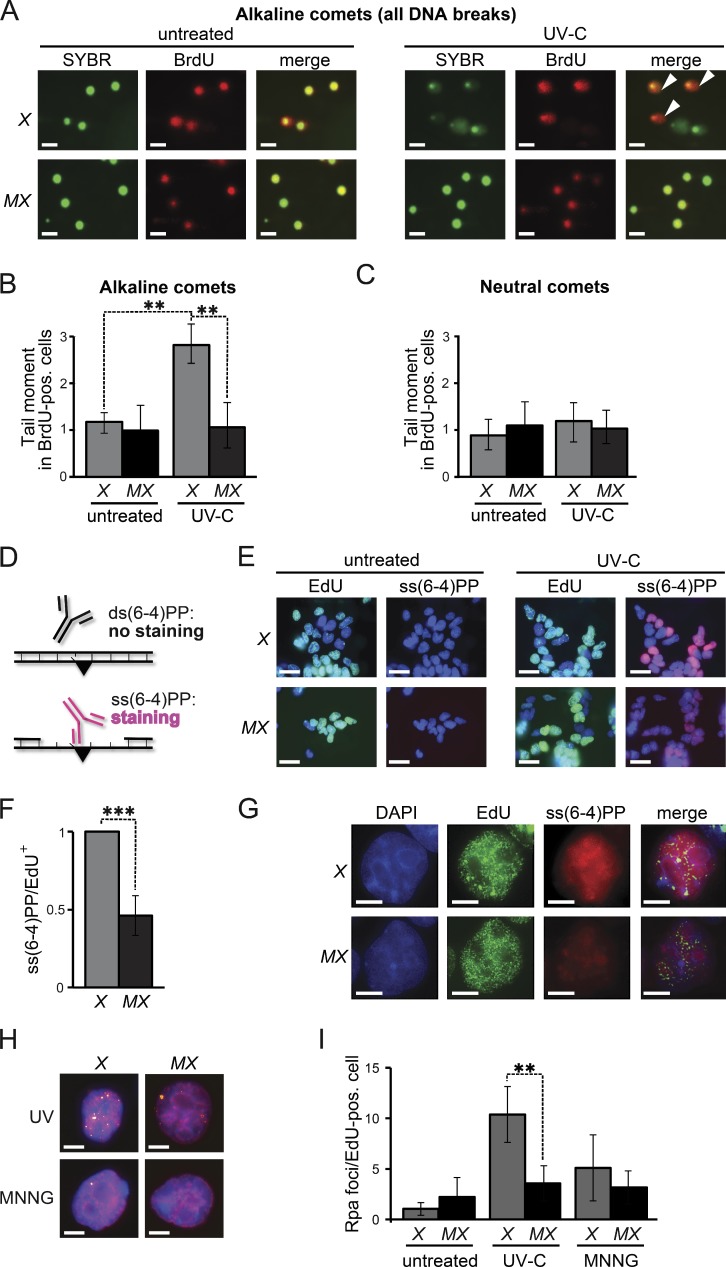
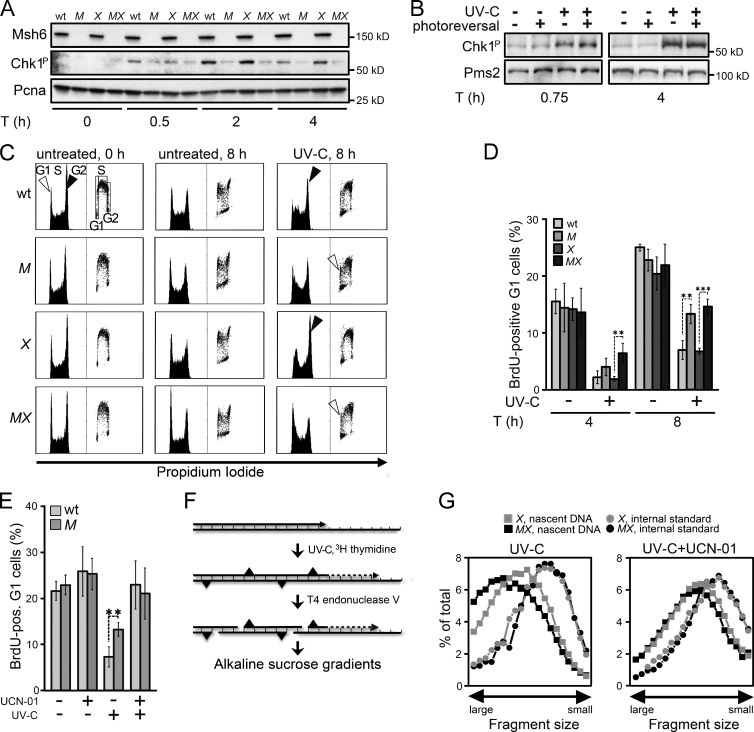
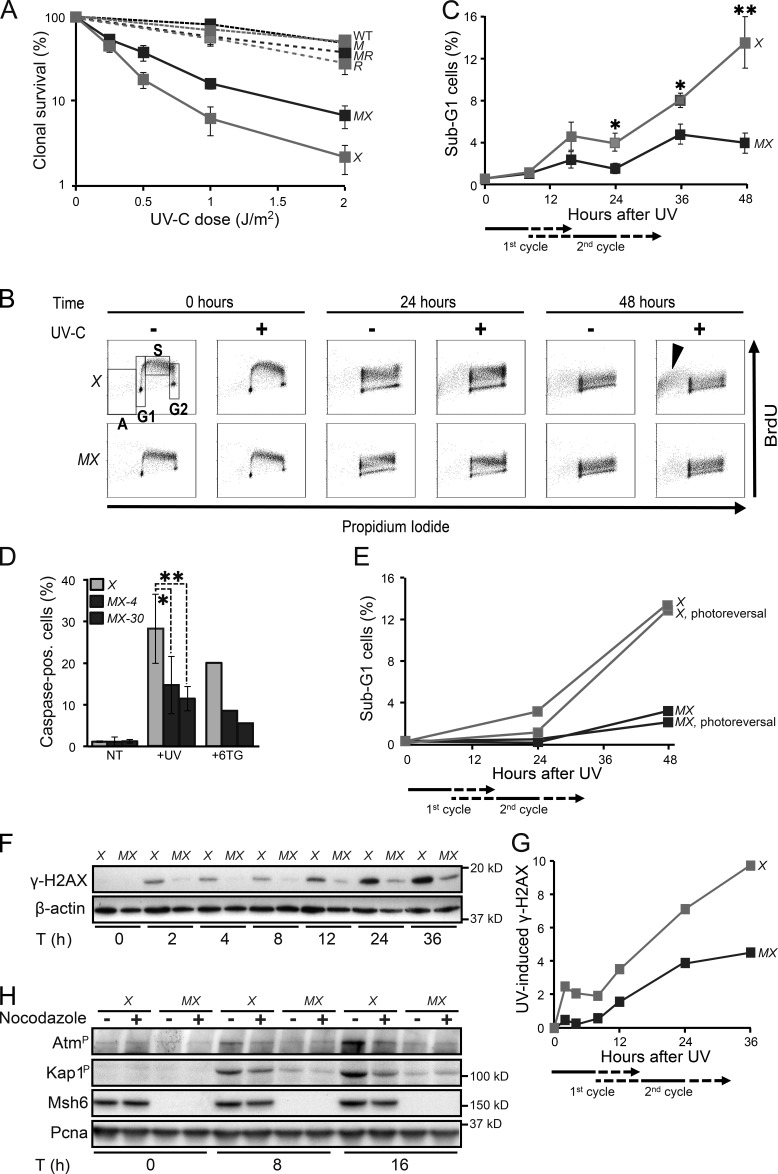
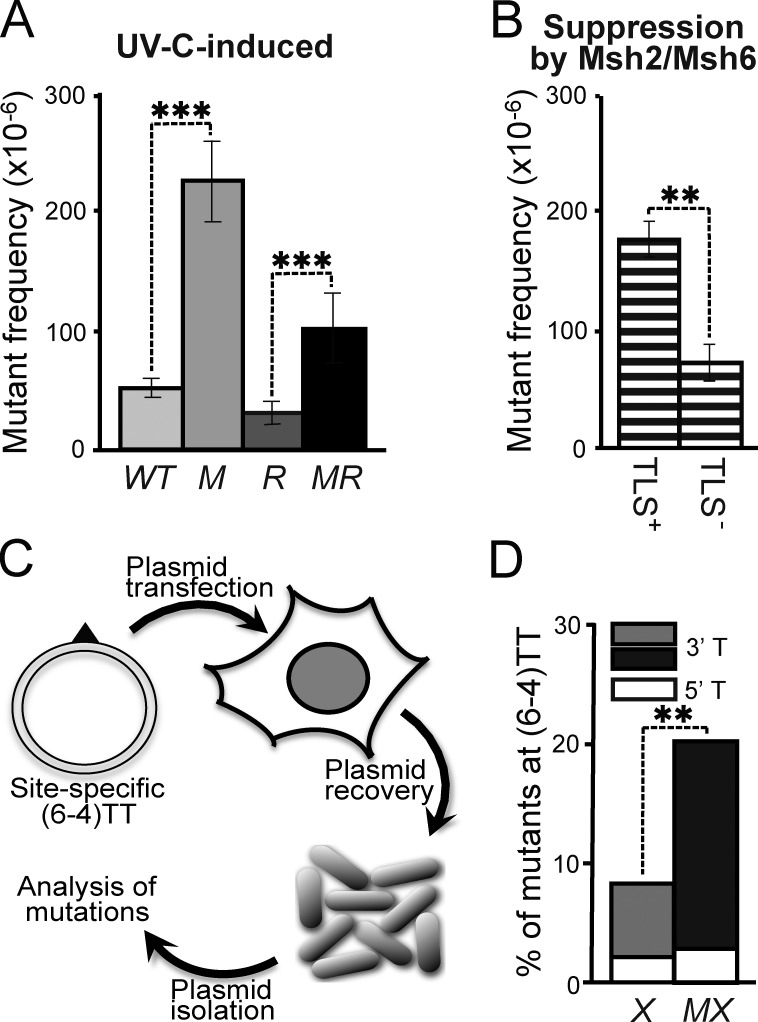
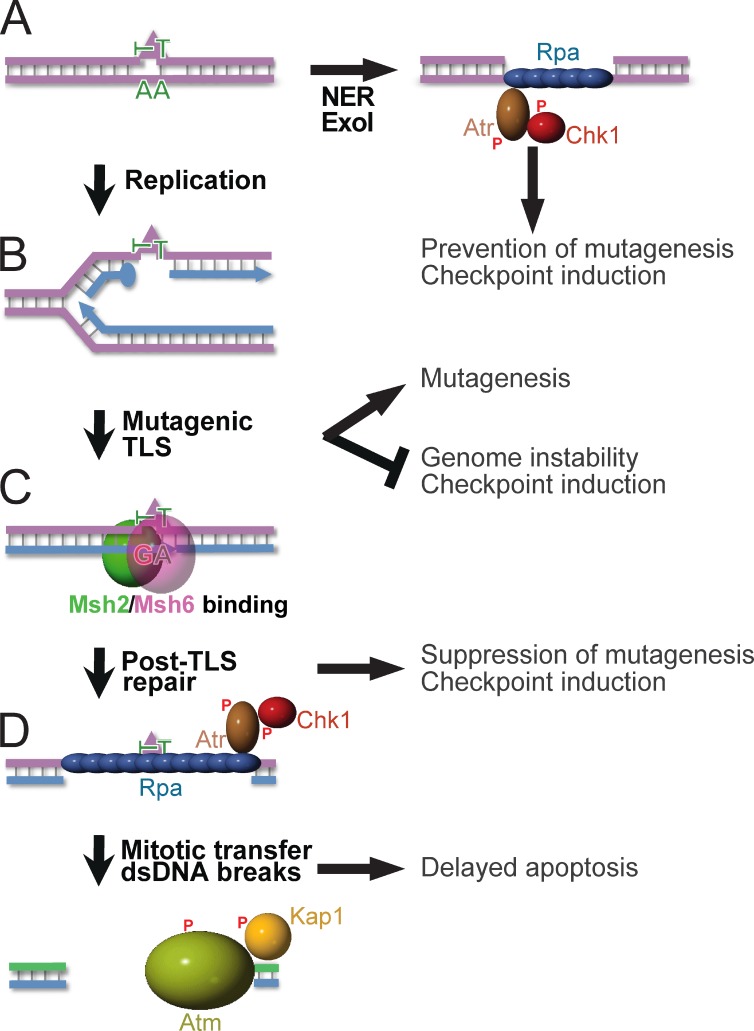
In addition to correcting mispaired nucleotides, DNA mismatch repair (MMR) proteins have been implicated in mutagenic, cell cycle, and apoptotic responses to agents that induce structurally aberrant nucleotide lesions. Here, we investigated the mechanistic basis for these responses by exposing cell lines with single or combined genetic defects in nucleotide excision repair (NER), postreplicative translesion synthesis (TLS), and MMR to low-dose ultraviolet light during S phase. Our data reveal that the MMR heterodimer Msh2/Msh6 mediates the excision of incorrect nucleotides that are incorporated by TLS opposite helix-distorting, noninstructive DNA photolesions. The resulting single-stranded DNA patches induce canonical Rpa-Atr-Chk1-mediated checkpoints and, in the next cell cycle, collapse to double-stranded DNA breaks that trigger apoptosis. In conclusion, a novel MMR-related DNA excision repair pathway controls TLS a posteriori, while initiating cellular responses to environmentally relevant densities of genotoxic lesions. These results may provide a rationale for the colorectal cancer tropism in Lynch syndrome, which is caused by inherited MMR gene defects.
© 2015 Tsaalbi-Shtylik et al.
Figures
Comment in
-
When mismatch repair met translesion synthesis.Cell Cycle. 2015 Aug 3;14(15):2377-8. doi: 10.1080/15384101.2015.1063288. Epub 2015 Jun 23. Cell Cycle. 2015. PMID: 26102362 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
