

So! What's aging? Is cardiovascular aging a disease?
- PMID: 25870157
- PMCID: PMC4532266
- DOI: 10.1016/j.yjmcc.2015.04.005
So! What's aging? Is cardiovascular aging a disease?
Abstract
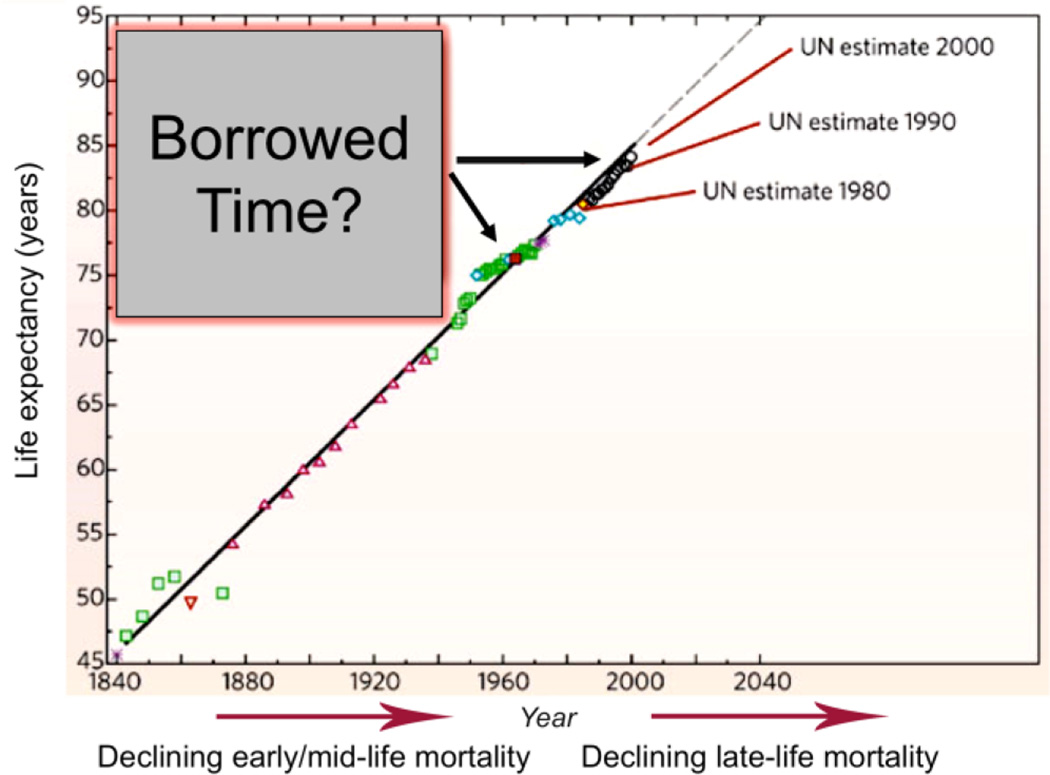
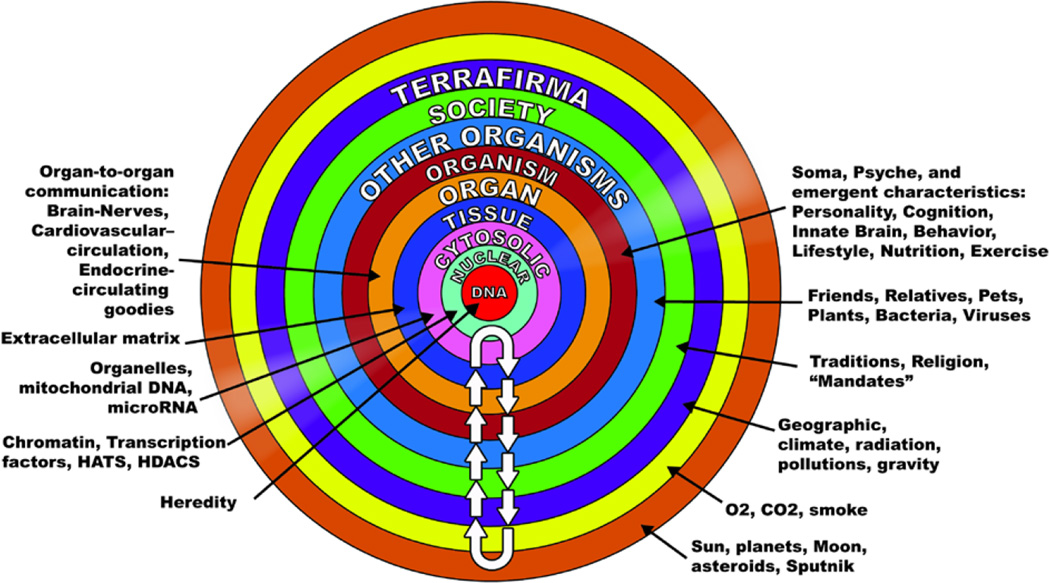
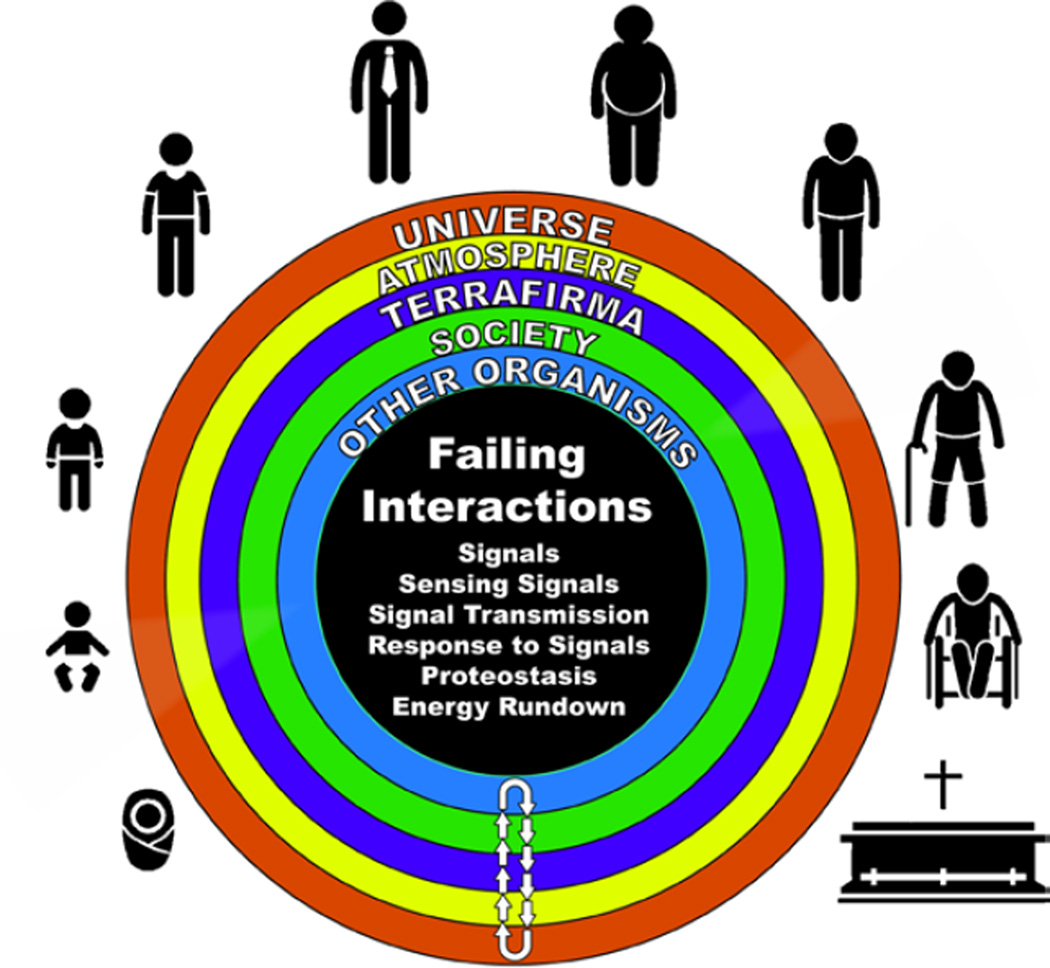
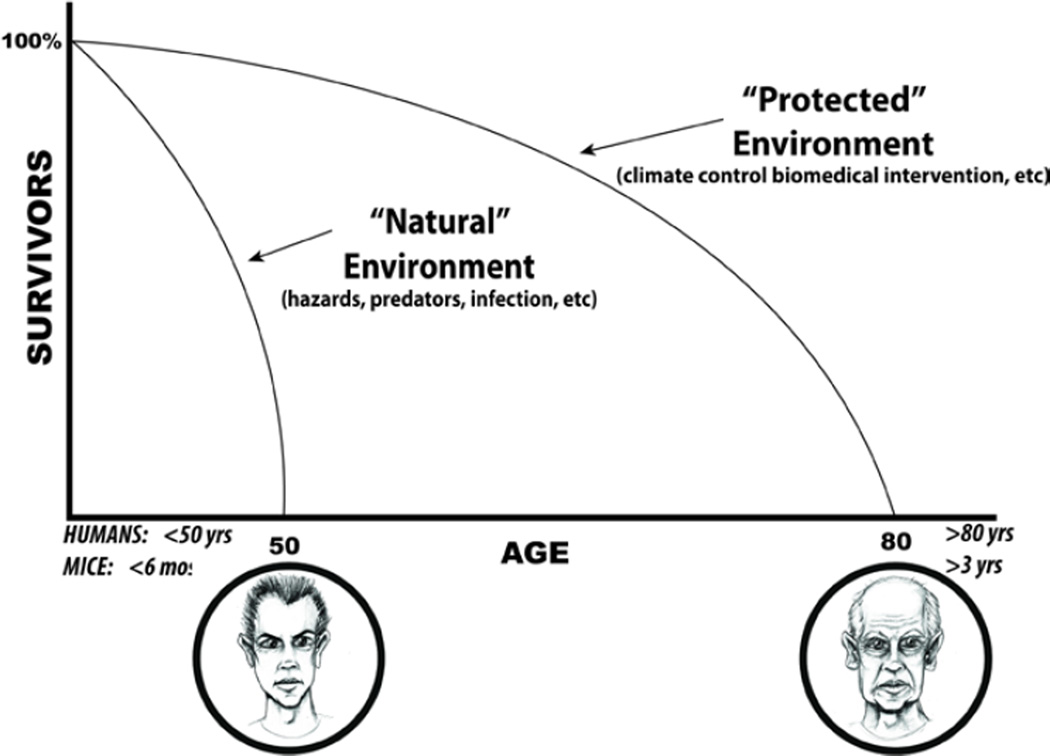
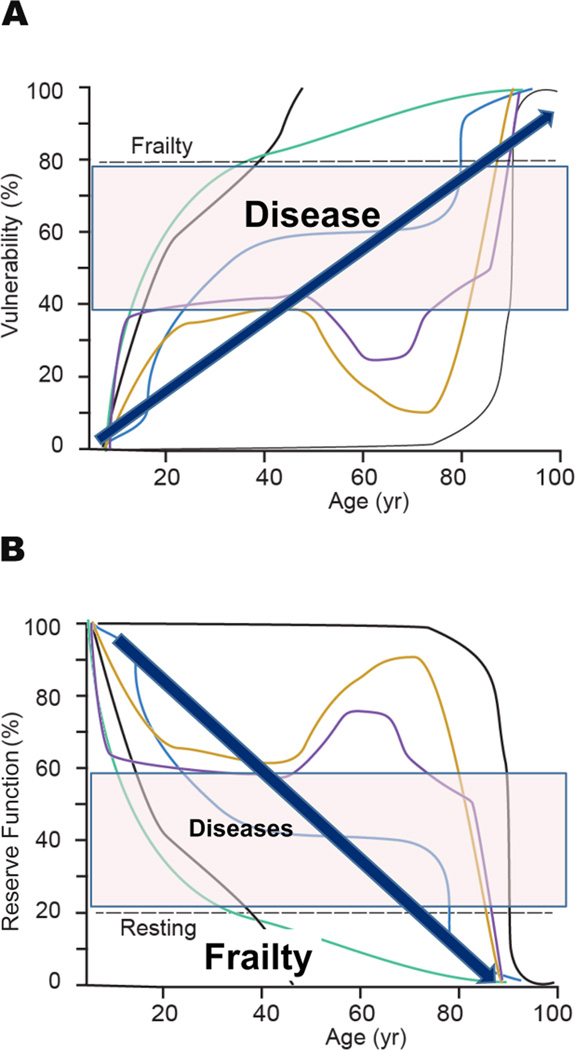
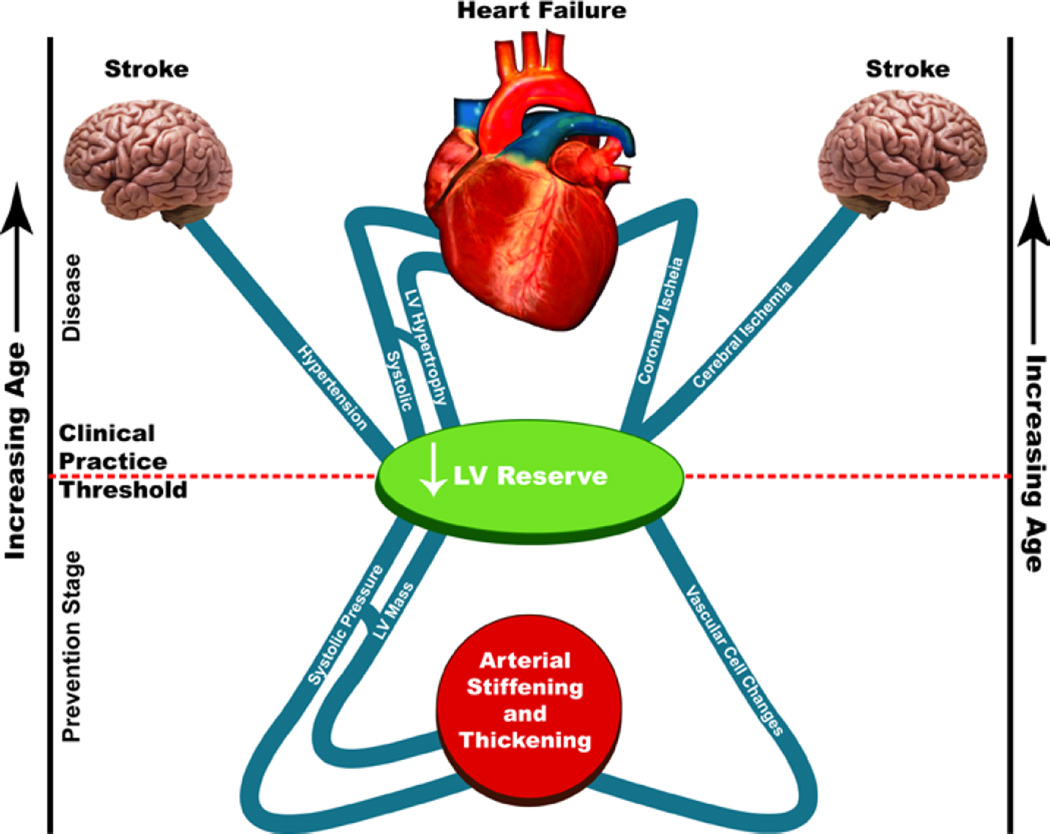
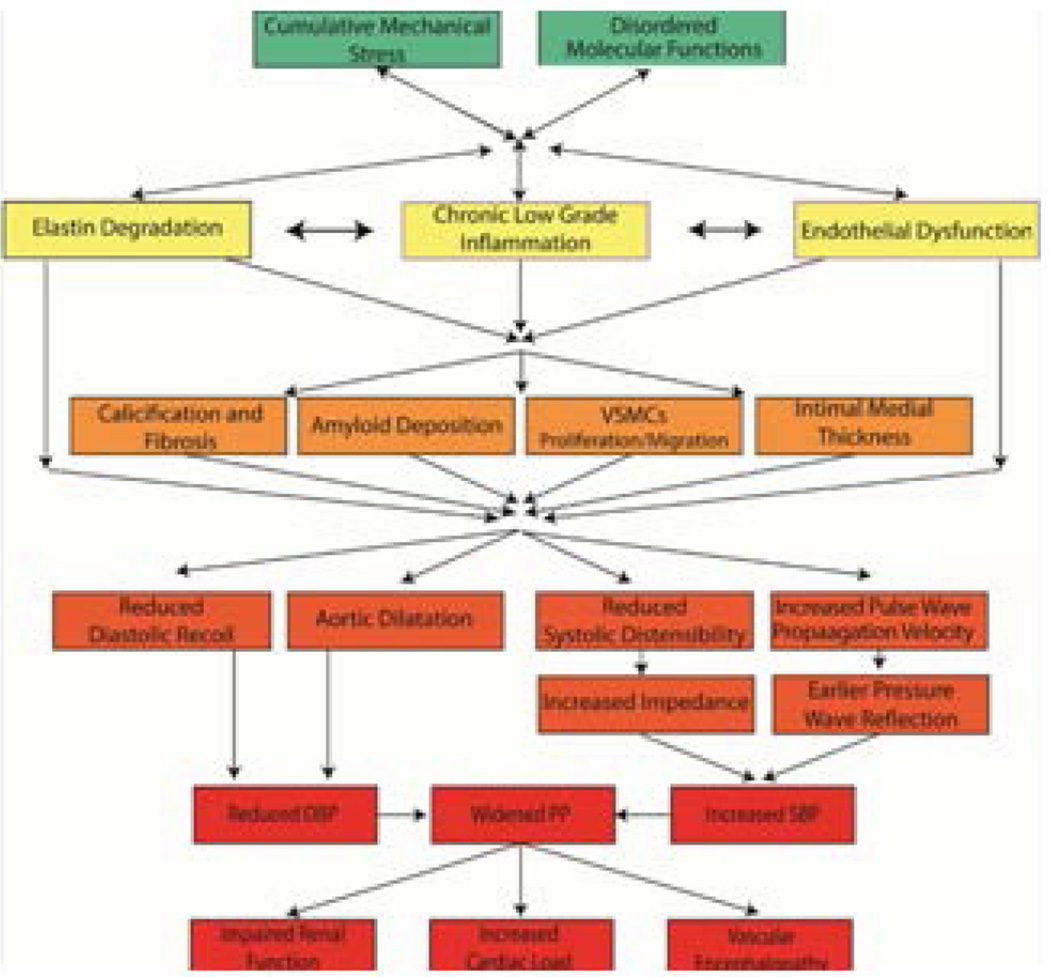
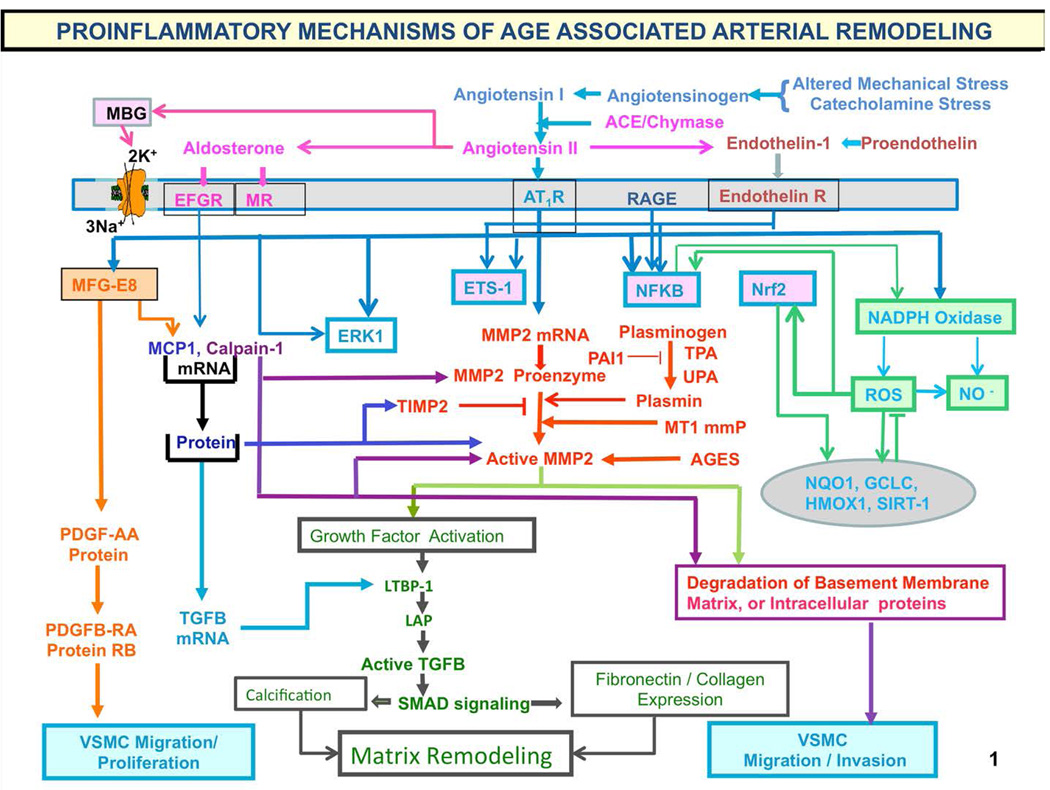
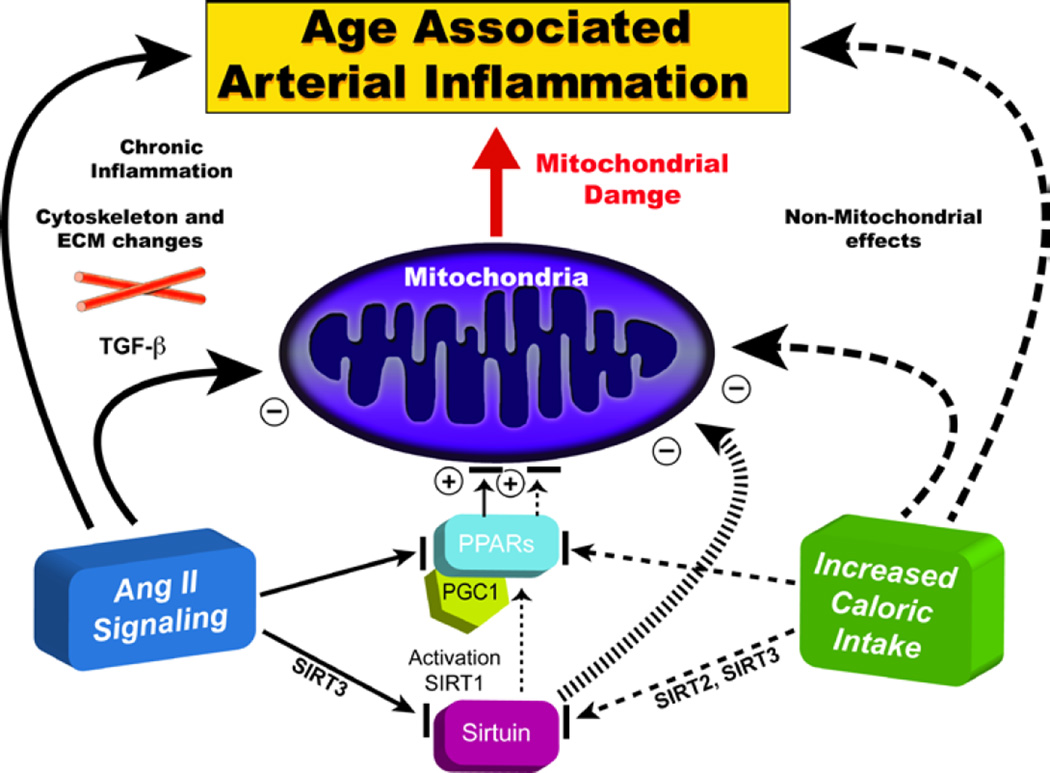
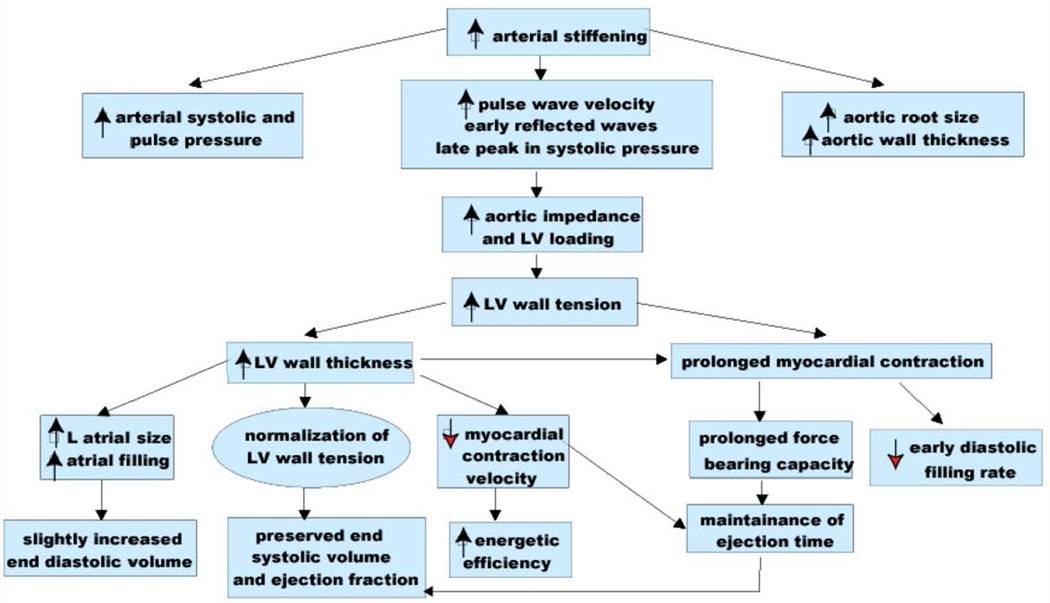
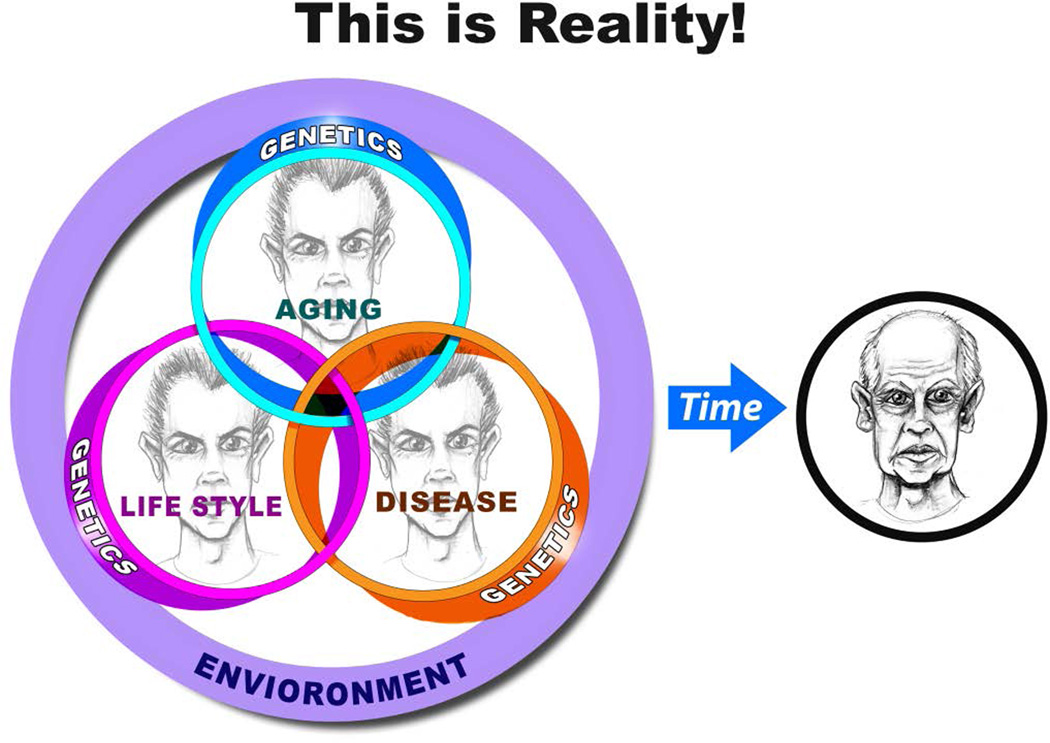
"Inside every old person is a young person wondering what happened." So, what is aging? Aging is a manifestation of progressive, time-dependent failure of molecular mechanisms that create disorder within a system of DNA and its environment (nuclear, cytosolic, tissue, organ, organism, other organisms, society, terra firma, atmosphere, universe). Continuous signaling, transmitted with different kinetics across each of these environments, confers a "mutual enslavement" that creates ordered functions among the components within the system. Accrual of this molecular disorder over time, i.e. during aging, causes progressive changes in the structure and function of the heart and arteries that are quite similar in humans, non-human primates, rabbits and rats that compromise cardiovascular reserve function, and confer a marked risk for incident cardiovascular disease. Nearly all aspects of signaling within the DNA environment system within the heart and arteries become disordered with advancing age: Signals change, as does sensing of the signals, transmission of signals and responses to signals, impaired cell renewal, changes in the proteome due to alterations in genomic transcription, mRNA translation, and proteostasis. The density of some molecules becomes reduced, and post-translational modifications, e.g. oxidation and nitration phosphorylation, lead to altered misfolding and disordered molecular interactions. The stoichiometry and kinetics of enzymatic and those reactions which underlie crucial cardiac and vascular cell functions and robust reserve mechanisms that remove damaged organelles and proteins deteriorate. The CV cells generate an inflammatory defense in an attempt to limit the molecular disorder. The resultant proinflammatory milieu is not executed by "professional" inflammatory cells (i.e. white blood cells), however, but by activation of renin-angiotensin-aldosterone endothelin signaling cascades that leads to endothelial and vascular smooth muscle and cardiac cells' phenotype shifts, resulting in production of inflammatory cytokines. Progressive molecular disorder within the heart and arteries over time leads to an excessive allostatic load on the CV system, that results in an increase and "overshoot" in the inflammatory defense signaling. This age-associated molecular disorder-induced inflammation that accrues in the heart and arteries does not, itself, cause clinical signs or symptoms of CVD. Clinical signs and symptoms of these CVDs begin to emerge, however, when the age-associated inflammation in the heart and arteries exceeds a threshold. Thus, an emerging school of thought is that accelerated age-associated alterations within the heart and arteries, per se, ought to be considered to be a type of CVD, because the molecular disorder and the inflammatory milieu it creates within the heart and arteries with advancing age are the roots of the pathophysiology of most cardiovascular diseases, e.g. athersclerosis and hypertension. Because many effects of aging on the CV system can be delayed or attenuated by changes in lifestyle, e.g. diet and exercise, or by presently available drugs, e.g. those that suppress Ang II signaling, CV aging is a promising frontier in preventive cardiology that is not only ripe for, but also in dire need of attention! There is an urgency to incorporate the concept of cardiovascular aging as a disease into clinical medicine. But, sadly, the reality of the age-associated molecular disorder within the heart and ateries has, for the most part, been kept outside of mainstream clinical medicine. This article is part of a Special Issue entitled CV Aging.
Published by Elsevier Ltd.
Figures



References
-
- Lakatta EG, Levy D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises. Part I: Aging Arteries: A “Set Up” for Vascular Disease. Circulation: New Frontiers. 2003;107:139–146. - PubMed
-
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update A report from the American Heart Association. Circulation. 2010;121:e1–e170.
-
- The World Health Report 1997—conquering, suffering, enriching humanity. Geneva: World Health Organization; - PubMed
-
- Kirkwood TB. A systematic look at an old problem. Nature. 2008 Feb 7;451(7179):644–647. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
