

Clustering molecular dynamics trajectories for optimizing docking experiments
- PMID: 25873944
- PMCID: PMC4385651
- DOI: 10.1155/2015/916240
Clustering molecular dynamics trajectories for optimizing docking experiments
Abstract
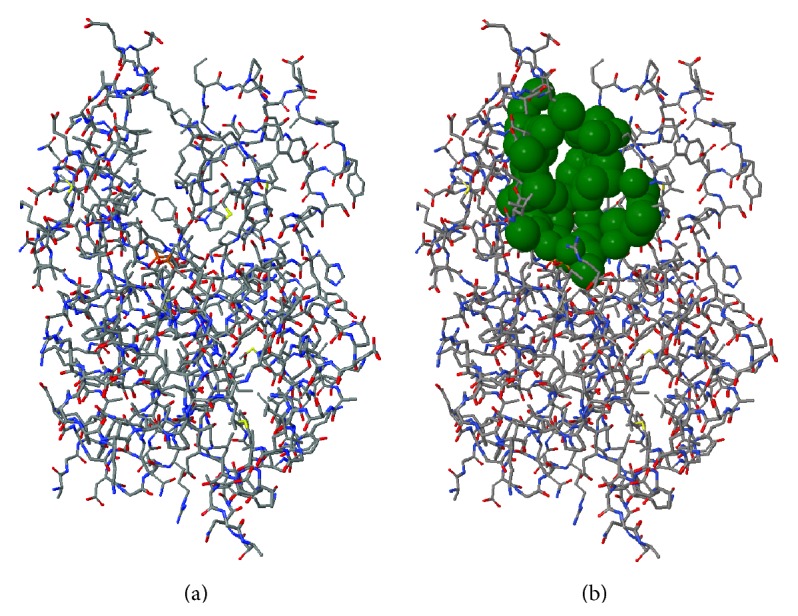
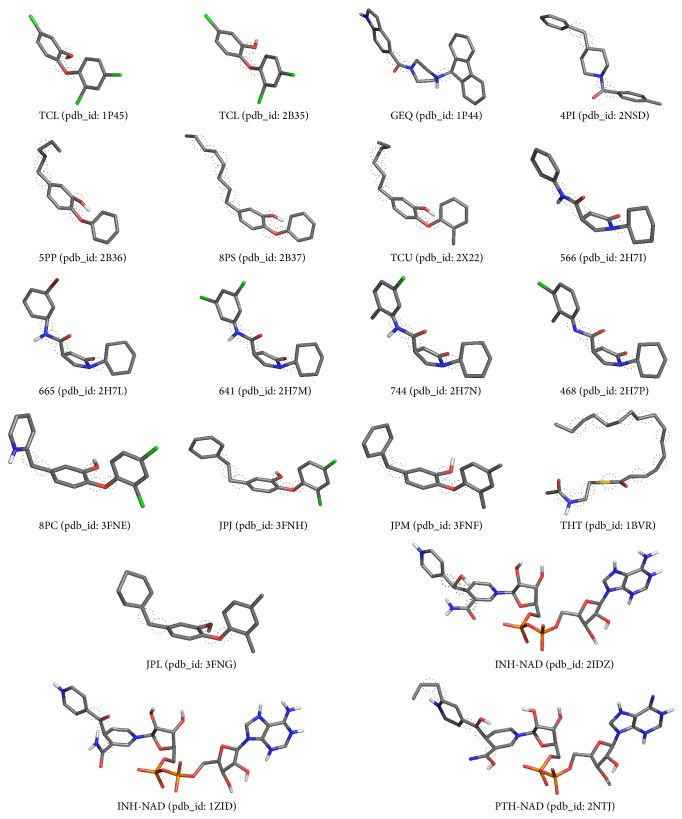
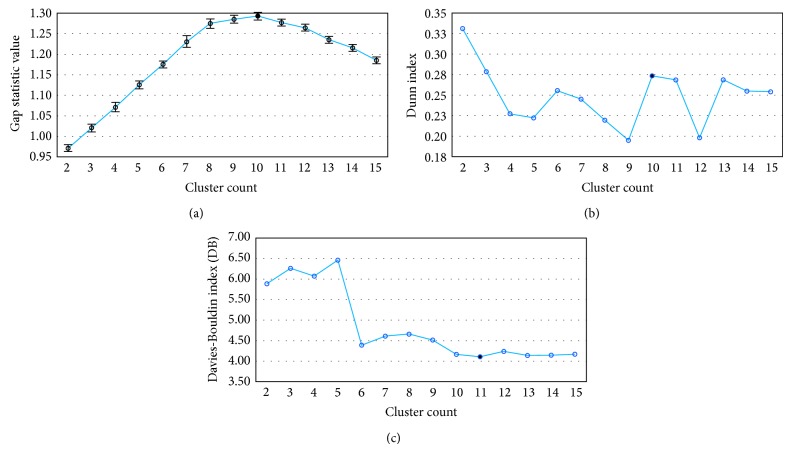
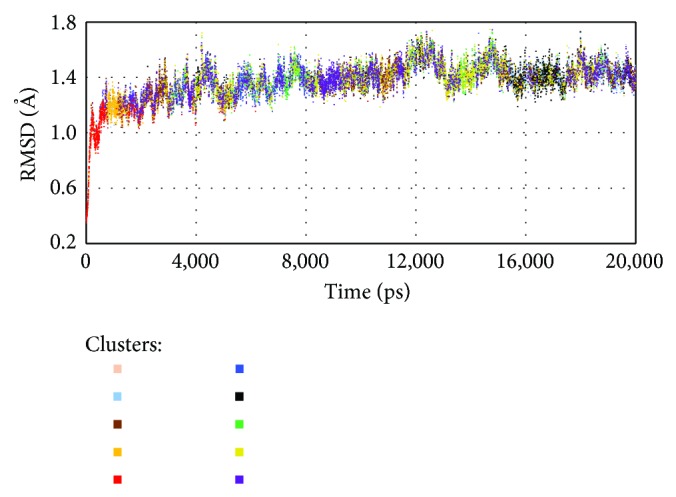
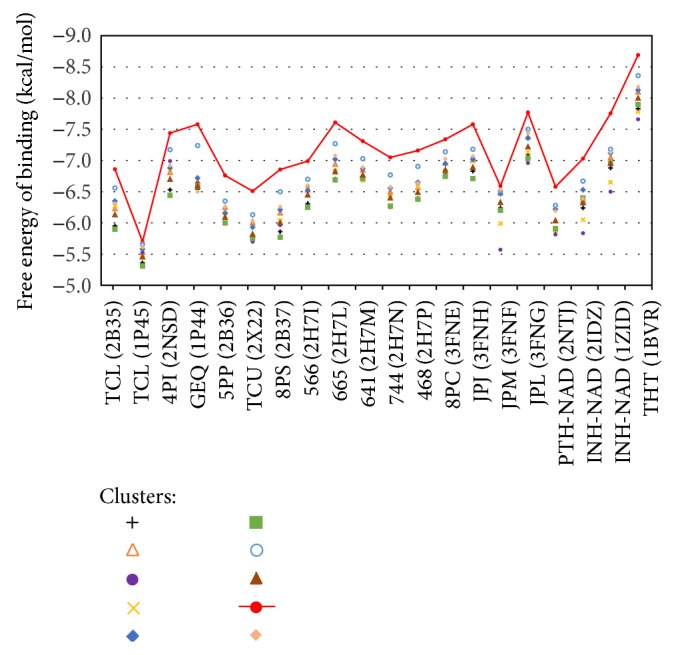
Molecular dynamics simulations of protein receptors have become an attractive tool for rational drug discovery. However, the high computational cost of employing molecular dynamics trajectories in virtual screening of large repositories threats the feasibility of this task. Computational intelligence techniques have been applied in this context, with the ultimate goal of reducing the overall computational cost so the task can become feasible. Particularly, clustering algorithms have been widely used as a means to reduce the dimensionality of molecular dynamics trajectories. In this paper, we develop a novel methodology for clustering entire trajectories using structural features from the substrate-binding cavity of the receptor in order to optimize docking experiments on a cloud-based environment. The resulting partition was selected based on three clustering validity criteria, and it was further validated by analyzing the interactions between 20 ligands and a fully flexible receptor (FFR) model containing a 20 ns molecular dynamics simulation trajectory. Our proposed methodology shows that taking into account features of the substrate-binding cavity as input for the k-means algorithm is a promising technique for accurately selecting ensembles of representative structures tailored to a specific ligand.
Figures
References
-
- Machado K. S., Winck A. T., Ruiz D. D., de Souza O. N. Mining flexible-receptor molecular docking data. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery. 2011;1(6):532–541. doi: 10.1002/widm.46. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
