

Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes
- PMID: 25874017
- PMCID: PMC4396570
- DOI: 10.1186/s13148-015-0064-6
Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes
Abstract
Background: Age-related changes in DNA methylation occurring in blood leukocytes during early childhood may reflect epigenetic maturation. We hypothesized that some of these changes involve gene networks of critical relevance in leukocyte biology and conducted a prospective study to elucidate the dynamics of DNA methylation. Serial blood samples were collected at 3, 6, 12, 24, 36, 48 and 60 months after birth in ten healthy girls born in Finland and participating in the Type 1 Diabetes Prediction and Prevention Study. DNA methylation was measured using the HumanMethylation450 BeadChip.
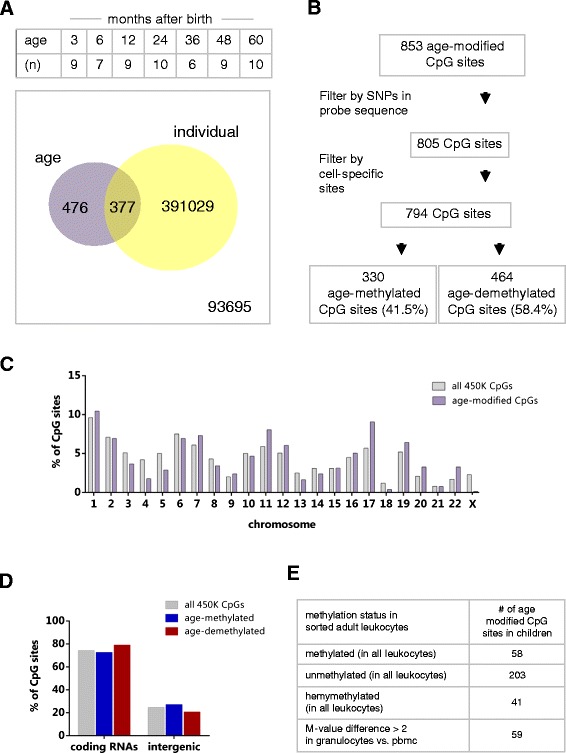
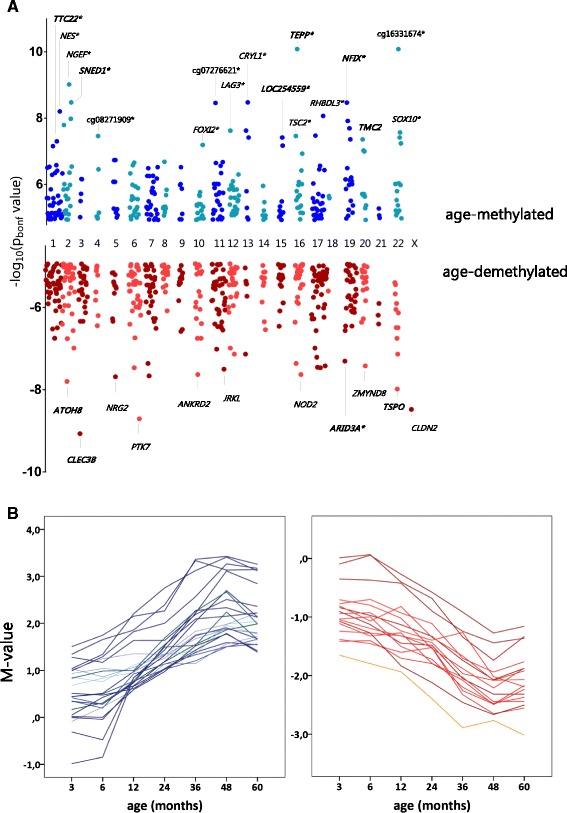
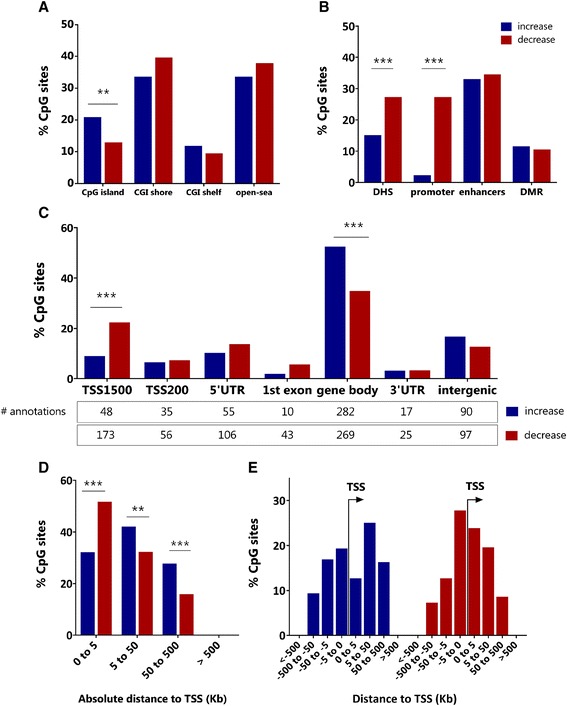
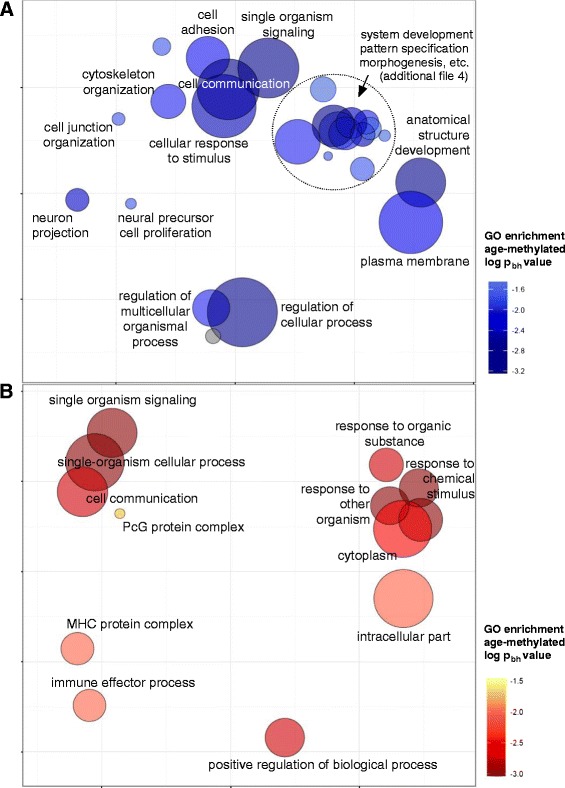
Results: After filtering for the presence of polymorphisms and cell-lineage-specific signatures, 794 CpG sites showed significant DNA methylation differences as a function of age in all children (41.6% age-methylated and 58.4% age-demethylated, Bonferroni-corrected P value <0.01). Age-methylated CpGs were more frequently located in gene bodies and within +5 to +50 kilobases (kb) of transcription start sites (TSS) and enriched in developmental, neuronal and plasma membrane genes. Age-demethylated CpGs were associated to promoters and DNAse-I hypersensitivity sites, located within -5 to +5 kb of the nearest TSS and enriched in genes related to immunity, antigen presentation, the polycomb-group protein complex and cytoplasm.
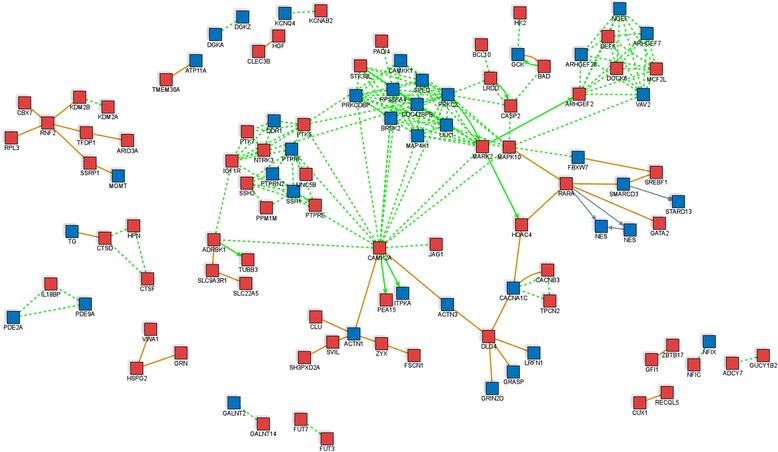
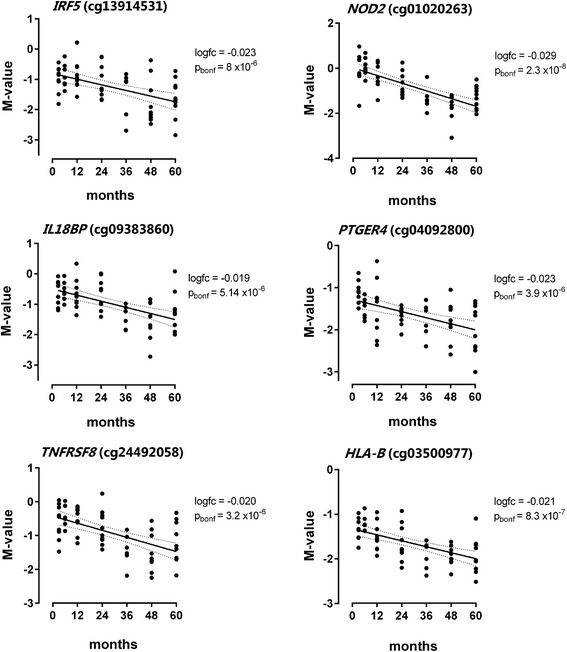
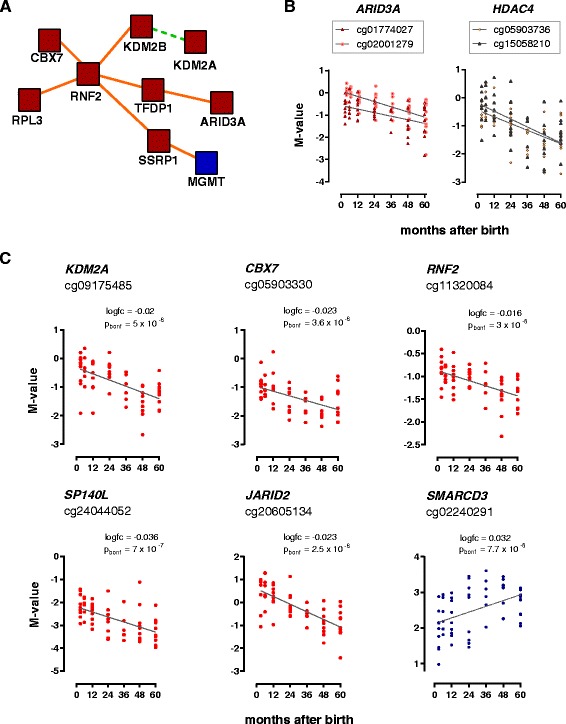
Conclusions: This study reveals that susceptibility loci for complex inflammatory diseases (for example, IRF5, NOD2, and PTGER4) and genes encoding histone modifiers and chromatin remodeling factors (for example, HDAC4, KDM2A, KDM2B, JARID2, ARID3A, and SMARCD3) undergo DNA methylation changes in leukocytes during early childhood. These results open new perspectives to understand leukocyte maturation and provide a catalogue of CpG sites that may need to be corrected for age effects when performing DNA methylation studies in children.
Keywords: Age-modified CpG; Childhood; DNA methylation; Genes; Leukocytes; Longitudinal.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
