

Systematic dissection of the sequence determinants of gene 3' end mediated expression control
- PMID: 25875337
- PMCID: PMC4398552
- DOI: 10.1371/journal.pgen.1005147
Systematic dissection of the sequence determinants of gene 3' end mediated expression control
Abstract
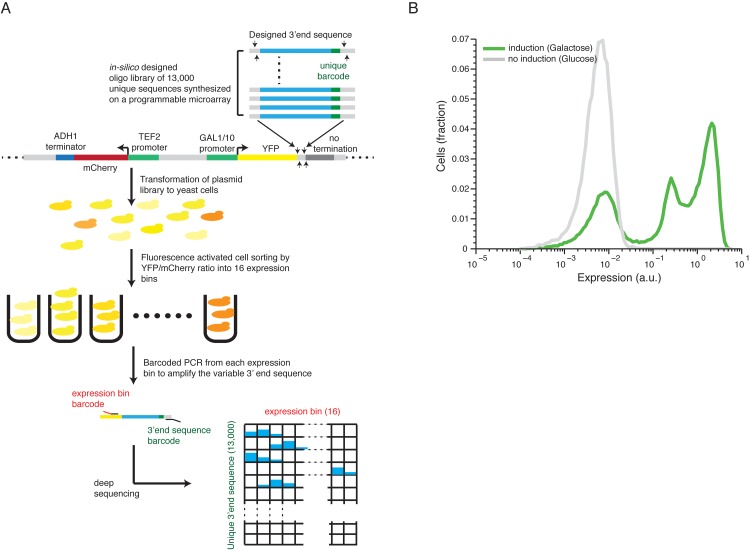
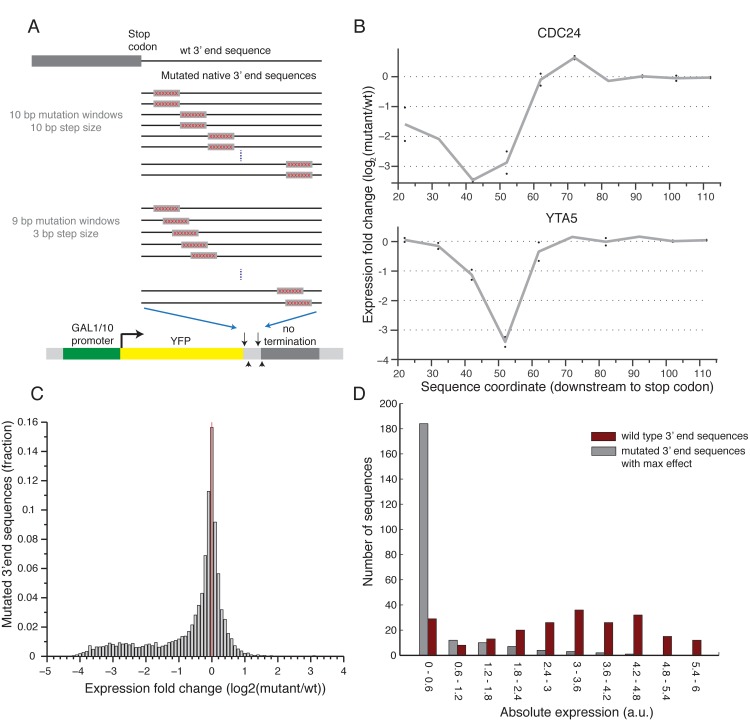
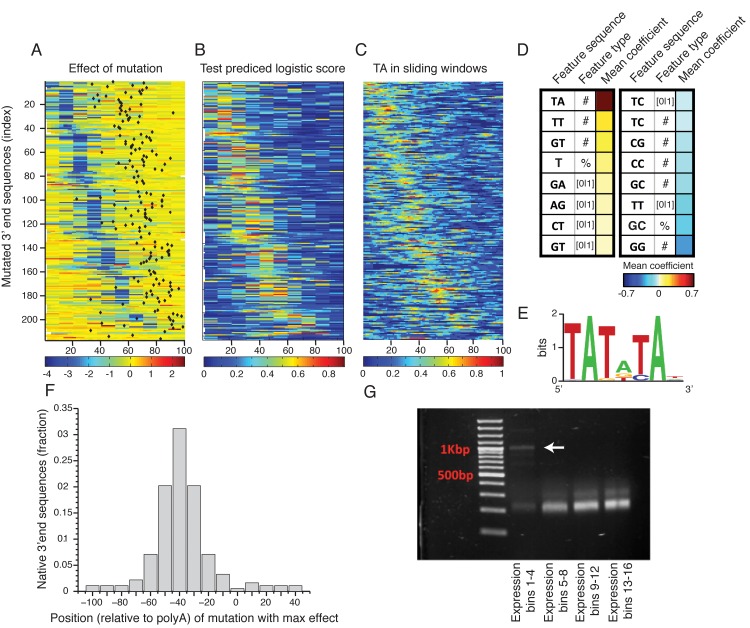
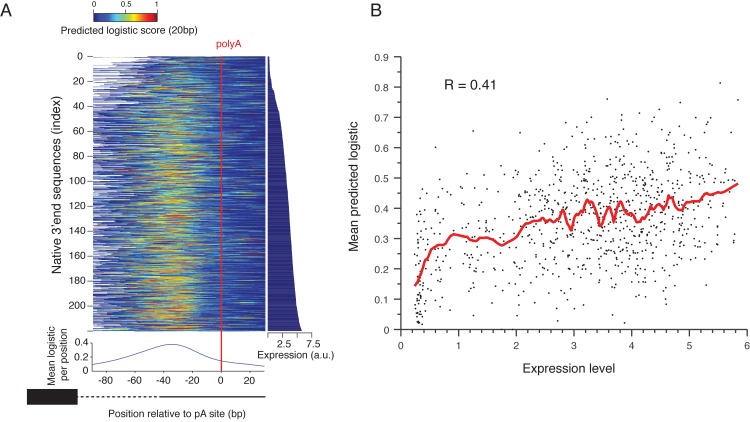
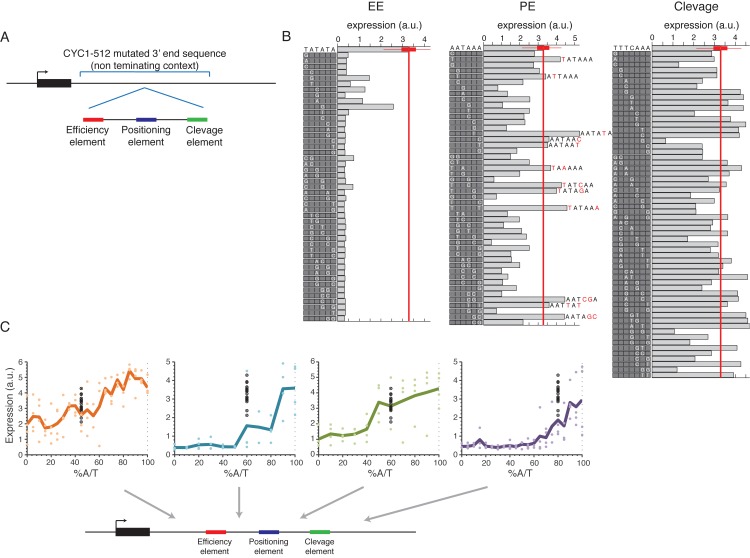
The 3'end genomic region encodes a wide range of regulatory process including mRNA stability, 3' end processing and translation. Here, we systematically investigate the sequence determinants of 3' end mediated expression control by measuring the effect of 13,000 designed 3' end sequence variants on constitutive expression levels in yeast. By including a high resolution scanning mutagenesis of more than 200 native 3' end sequences in this designed set, we found that most mutations had only a mild effect on expression, and that the vast majority (~90%) of strongly effecting mutations localized to a single positive TA-rich element, similar to a previously described 3' end processing efficiency element, and resulted in up to ten-fold decrease in expression. Measurements of 3' UTR lengths revealed that these mutations result in mRNAs with aberrantly long 3'UTRs, confirming the role for this element in 3' end processing. Interestingly, we found that other sequence elements that were previously described in the literature to be part of the polyadenylation signal had a minor effect on expression. We further characterize the sequence specificities of the TA-rich element using additional synthetic 3' end sequences and show that its activity is sensitive to single base pair mutations and strongly depends on the A/T content of the surrounding sequences. Finally, using a computational model, we show that the strength of this element in native 3' end sequences can explain some of their measured expression variability (R = 0.41). Together, our results emphasize the importance of efficient 3' end processing for endogenous protein levels and contribute to an improved understanding of the sequence elements involved in this process.
Conflict of interest statement
ZY is an employee of and owns stocks in Agilent Technologies. Other authors have declared that no competing interests exist.
Figures
References
-
- Hammell CM, Gross S, Zenklusen D, Heath C V, Stutz F, Moore C, et al. Coupling of termination, 3’ processing, and mRNA export. Mol Cell Biol. 2002;22: 6441–57. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=135649&tool=pm... - PMC - PubMed
-
- Birse CE, Minvielle-Sebastia L, Lee BA, Keller W, Proudfoot NJ. Coupling termination of transcription to messenger RNA maturation in yeast. Science. 1998;280: 298–301. Available: http://www.ncbi.nlm.nih.gov/pubmed/9535662 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
