

Whole-genome sequencing in outbreak analysis
- PMID: 25876885
- PMCID: PMC4399107
- DOI: 10.1128/CMR.00075-13
Whole-genome sequencing in outbreak analysis
Abstract
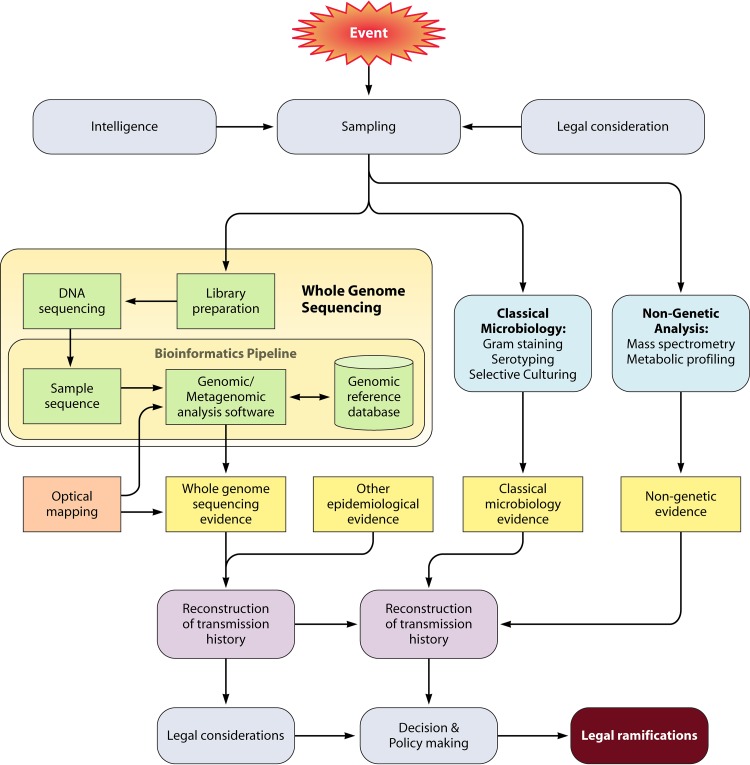
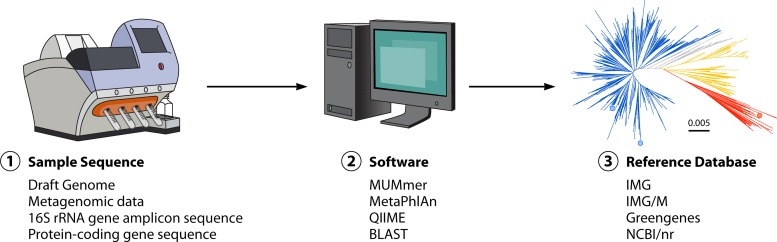
In addition to the ever-present concern of medical professionals about epidemics of infectious diseases, the relative ease of access and low cost of obtaining, producing, and disseminating pathogenic organisms or biological toxins mean that bioterrorism activity should also be considered when facing a disease outbreak. Utilization of whole-genome sequencing (WGS) in outbreak analysis facilitates the rapid and accurate identification of virulence factors of the pathogen and can be used to identify the path of disease transmission within a population and provide information on the probable source. Molecular tools such as WGS are being refined and advanced at a rapid pace to provide robust and higher-resolution methods for identifying, comparing, and classifying pathogenic organisms. If these methods of pathogen characterization are properly applied, they will enable an improved public health response whether a disease outbreak was initiated by natural events or by accidental or deliberate human activity. The current application of next-generation sequencing (NGS) technology to microbial WGS and microbial forensics is reviewed.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures





References
-
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. 2012. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi: 10.1186/1471-2164-13-341. - DOI - PMC - PubMed
-
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11:R119. doi: 10.1186/gb-2010-11-12-r119. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
