

Stable Phenotypic Changes of the Host T Cells Are Essential to the Long-Term Stability of Latent HIV-1 Infection
- PMID: 25878110
- PMCID: PMC4468477
- DOI: 10.1128/JVI.00571-15
Stable Phenotypic Changes of the Host T Cells Are Essential to the Long-Term Stability of Latent HIV-1 Infection
Abstract
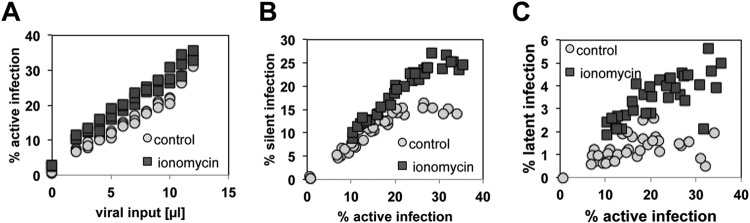
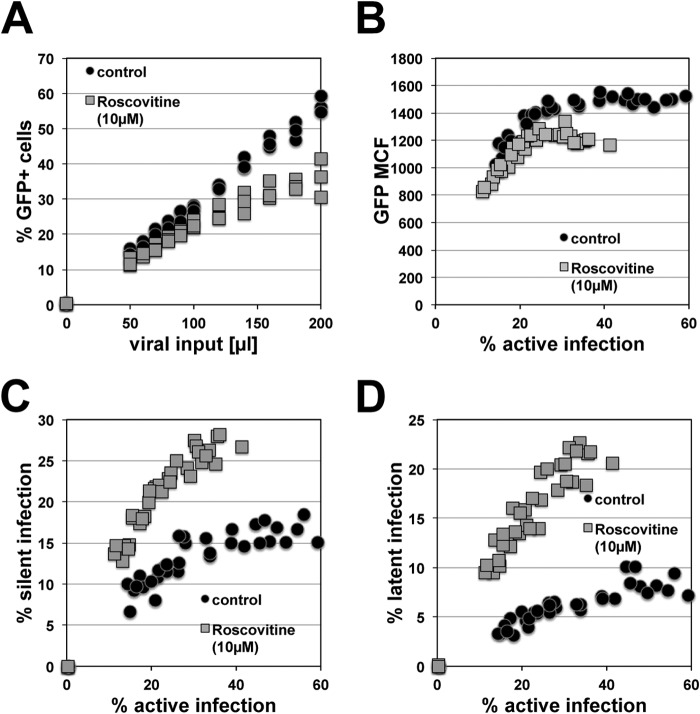
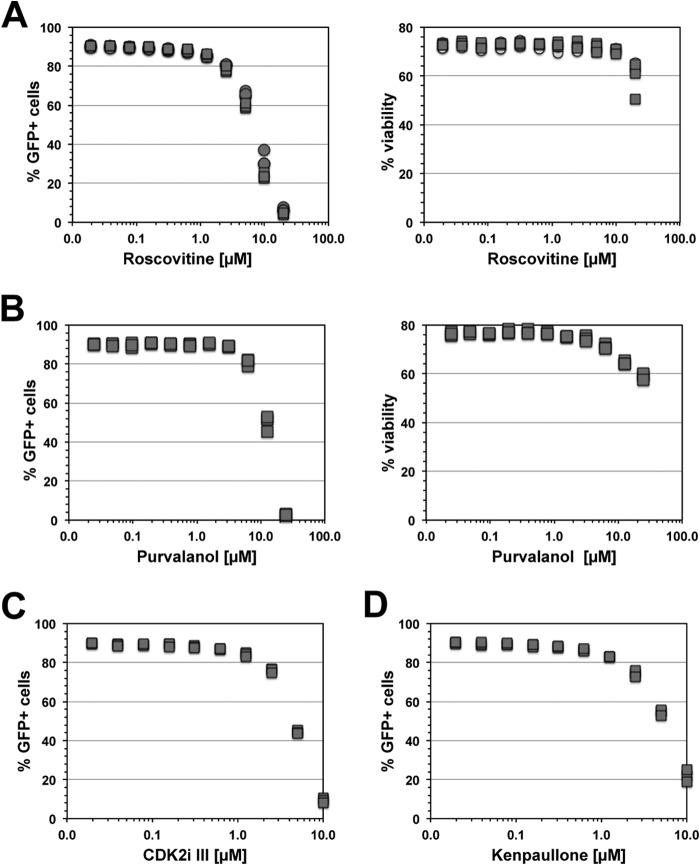
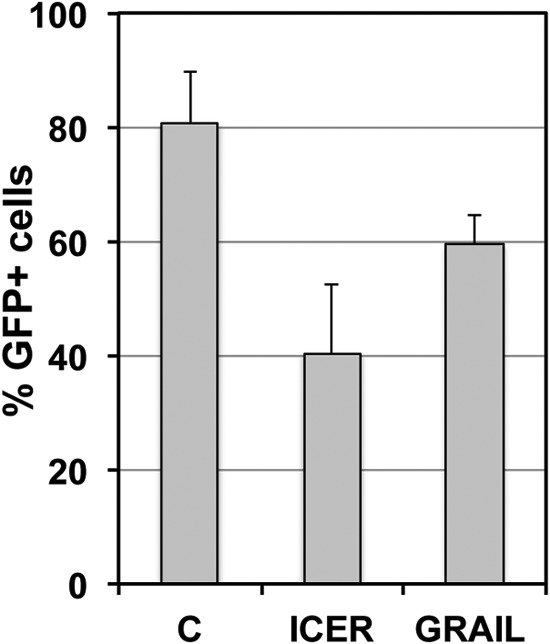
The extreme stability of the latent HIV-1 reservoir in the CD4(+) memory T cell population prevents viral eradication with current antiretroviral therapy. It has been demonstrated that homeostatic T cell proliferation and clonal expansion of latently infected T cells due to viral integration into specific genes contribute to this extraordinary reservoir stability. Nevertheless, given the constant exposure of the memory T cell population to specific antigen or bystander activation, this reservoir stability seems remarkable, unless it is assumed that latent HIV-1 resides exclusively in memory T cells that recognize rare antigens. Another explanation for the stability of the reservoir could be that the latent HIV-1 reservoir is associated with an unresponsive T cell phenotype. We demonstrate here that host cells of latent HIV-1 infection events were functionally altered in ways that are consistent with the idea of an anergic, unresponsive T cell phenotype. Manipulations that induced or mimicked an anergic T cell state promoted latent HIV-1 infection. Kinome analysis data reflected this altered host cell phenotype at a system-wide level and revealed how the stable kinase activity changes networked to stabilize latent HIV-1 infection. Protein-protein interaction networks generated from kinome data could further be used to guide targeted genetic or pharmacological manipulations that alter the stability of latent HIV-1 infection. In summary, our data demonstrate that stable changes to the signal transduction and transcription factor network of latently HIV-1 infected host cells are essential to the ability of HIV-1 to establish and maintain latent HIV-1 infection status.
Importance: The extreme stability of the latent HIV-1 reservoir allows the infection to persist for the lifetime of a patient, despite completely suppressive antiretroviral therapy. This extreme reservoir stability is somewhat surprising, since the latently HIV-1 infected CD4(+) memory T cells that form the structural basis of the viral reservoir should be exposed to cognate antigen over time. Antigen exposure would trigger a recall response and should deplete the reservoir, likely over a relatively short period. Our data demonstrate that stable and system-wide phenotypic changes to host cells are a prerequisite for the establishment and maintenance of latent HIV-1 infection events. The changes observed are consistent with an unresponsive, anergy-like T cell phenotype of latently HIV-1 infected host cells. An anergy-like, unresponsive state of the host cells of latent HIV-1 infection events would explain the stability of the HIV-1 reservoir in the face of continuous antigen exposure.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy.Annu Rev Immunol. 2000;18:665-708. doi: 10.1146/annurev.immunol.18.1.665. Annu Rev Immunol. 2000. PMID: 10837072 Review.
-
A stable latent reservoir for HIV-1 in resting CD4(+) T lymphocytes in infected children.J Clin Invest. 2000 Apr;105(7):995-1003. doi: 10.1172/JCI9006. J Clin Invest. 2000. PMID: 10749578 Free PMC article.
-
Stochastic population switch may explain the latent reservoir stability and intermittent viral blips in HIV patients on suppressive therapy.J Theor Biol. 2014 Nov 7;360:137-148. doi: 10.1016/j.jtbi.2014.06.042. Epub 2014 Jul 10. J Theor Biol. 2014. PMID: 25016044
-
CD161+ CD4+ T Cells Harbor Clonally Expanded Replication-Competent HIV-1 in Antiretroviral Therapy-Suppressed Individuals.mBio. 2019 Oct 8;10(5):e02121-19. doi: 10.1128/mBio.02121-19. mBio. 2019. PMID: 31594817 Free PMC article.
-
The Latent Reservoir for HIV-1: How Immunologic Memory and Clonal Expansion Contribute to HIV-1 Persistence.J Immunol. 2016 Jul 15;197(2):407-17. doi: 10.4049/jimmunol.1600343. J Immunol. 2016. PMID: 27382129 Free PMC article. Review.
Cited by
-
CD4+ T cells from HIV-1 patients with impaired Th1 effector responses to Mycobacterium tuberculosis exhibit diminished histone and nucleoprotein signatures.Clin Immunol. 2017 Aug;181:16-23. doi: 10.1016/j.clim.2017.05.018. Epub 2017 May 25. Clin Immunol. 2017. PMID: 28552470 Free PMC article.
-
HIV-1 Reservoirs During Suppressive Therapy.Trends Microbiol. 2016 May;24(5):345-355. doi: 10.1016/j.tim.2016.01.006. Epub 2016 Feb 12. Trends Microbiol. 2016. PMID: 26875617 Free PMC article. Review.
-
Nucleolar protein NOP2/NSUN1 suppresses HIV-1 transcription and promotes viral latency by competing with Tat for TAR binding and methylation.PLoS Pathog. 2020 Mar 16;16(3):e1008430. doi: 10.1371/journal.ppat.1008430. eCollection 2020 Mar. PLoS Pathog. 2020. PMID: 32176734 Free PMC article.
-
Peering into the HIV reservoir.Rev Med Virol. 2018 Jul;28(4):e1981. doi: 10.1002/rmv.1981. Epub 2018 May 9. Rev Med Virol. 2018. PMID: 29744964 Free PMC article. Review.
-
The landscape of alternative splicing in HIV-1 infected CD4 T-cells.BMC Med Genomics. 2020 Apr 3;13(Suppl 5):38. doi: 10.1186/s12920-020-0680-7. BMC Med Genomics. 2020. PMID: 32241262 Free PMC article.
References
-
- Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi:10.1038/387183a0. - DOI - PubMed
-
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi:10.1038/8394. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
