

Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease
- PMID: 25878292
- PMCID: PMC4397611
- DOI: 10.1523/JNEUROSCI.2552-14.2015
Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer's disease
Abstract
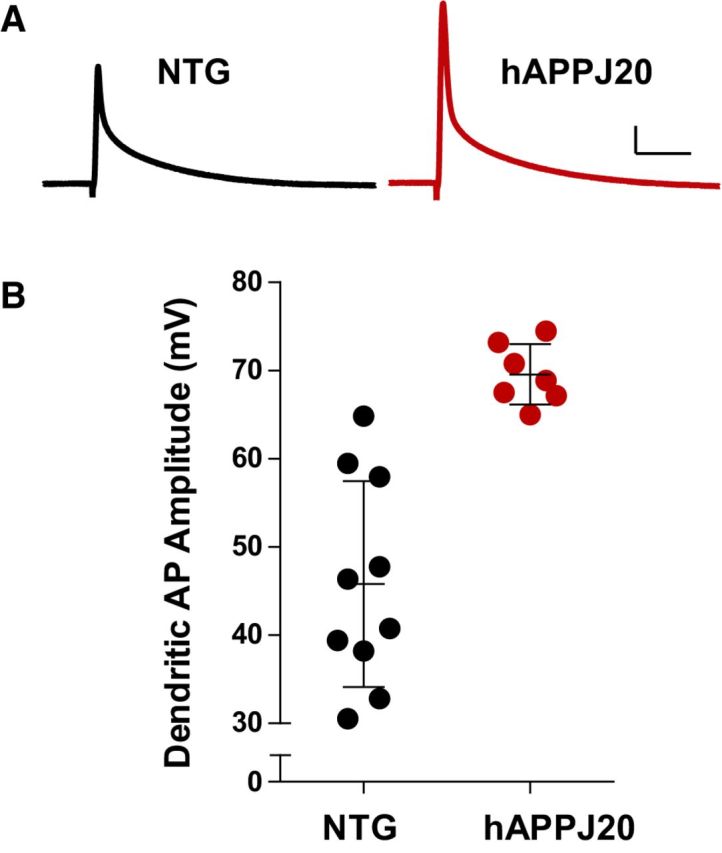
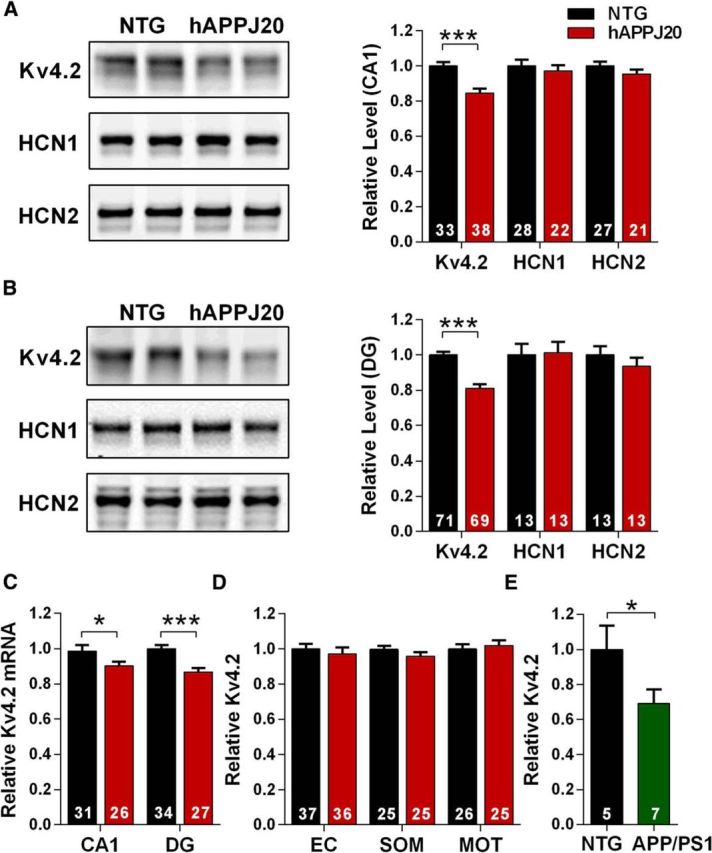
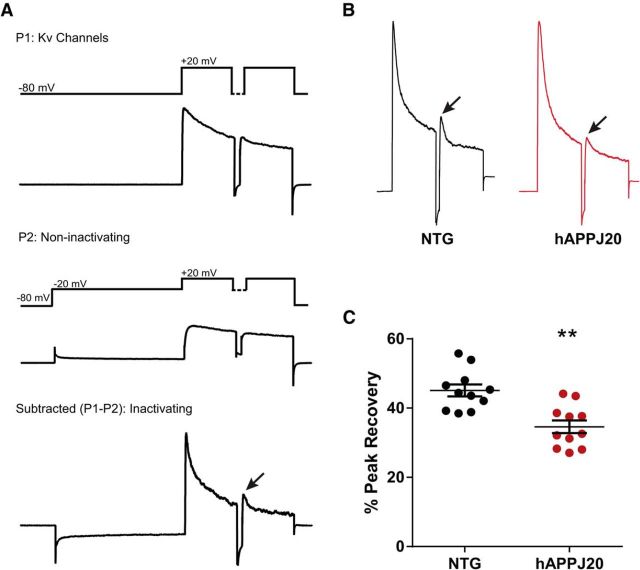
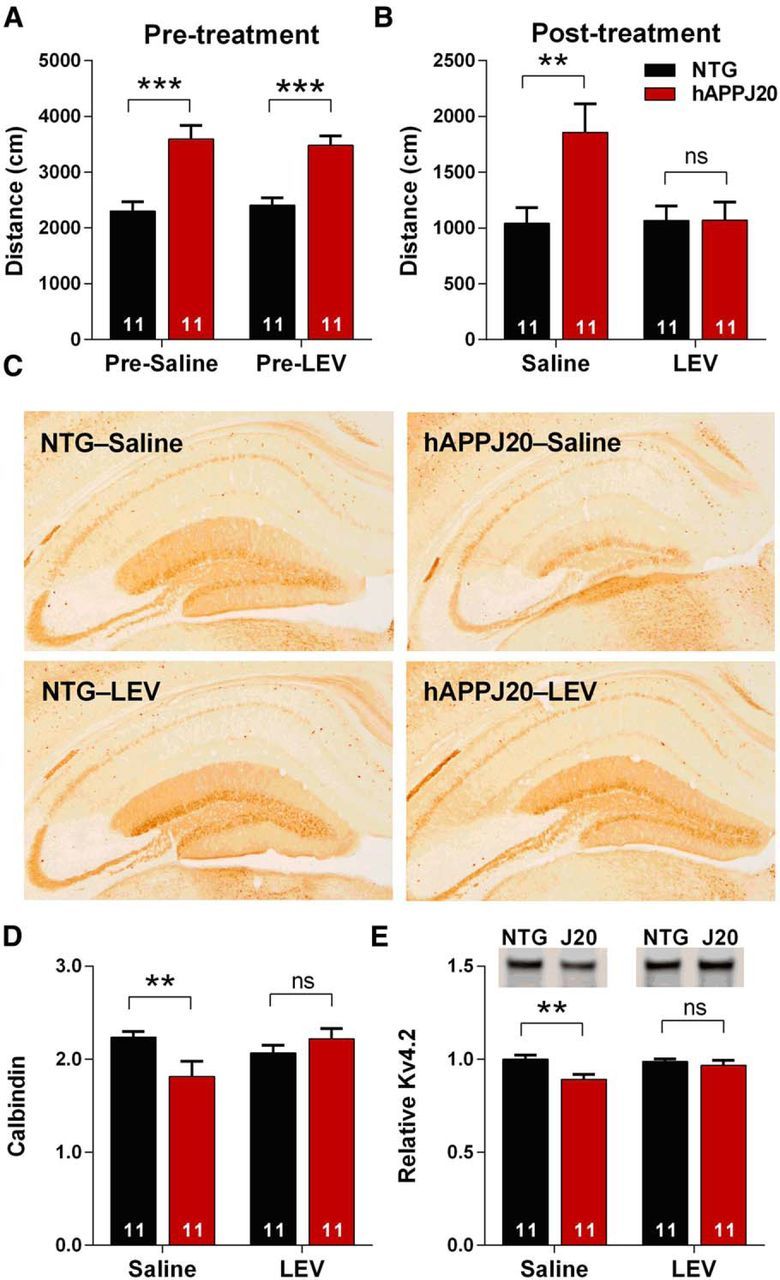
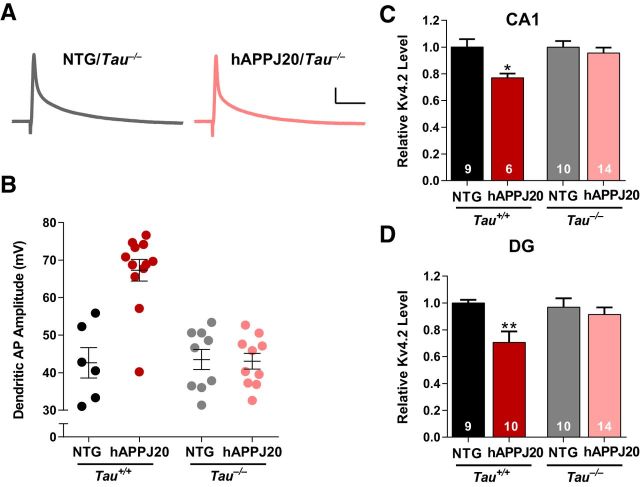
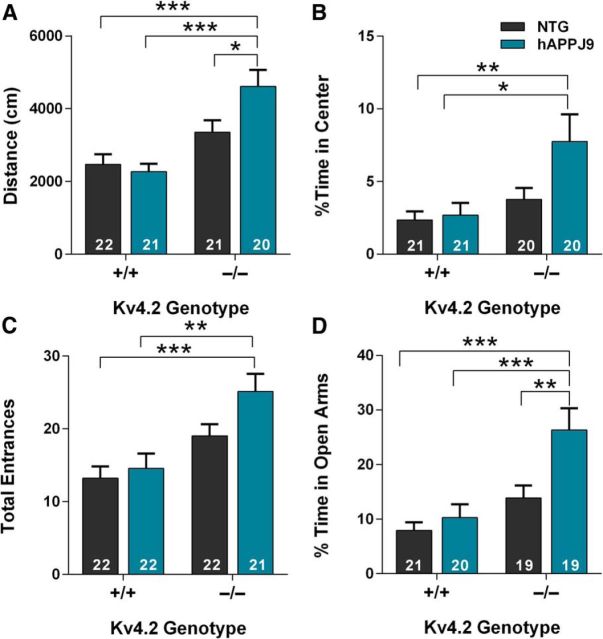
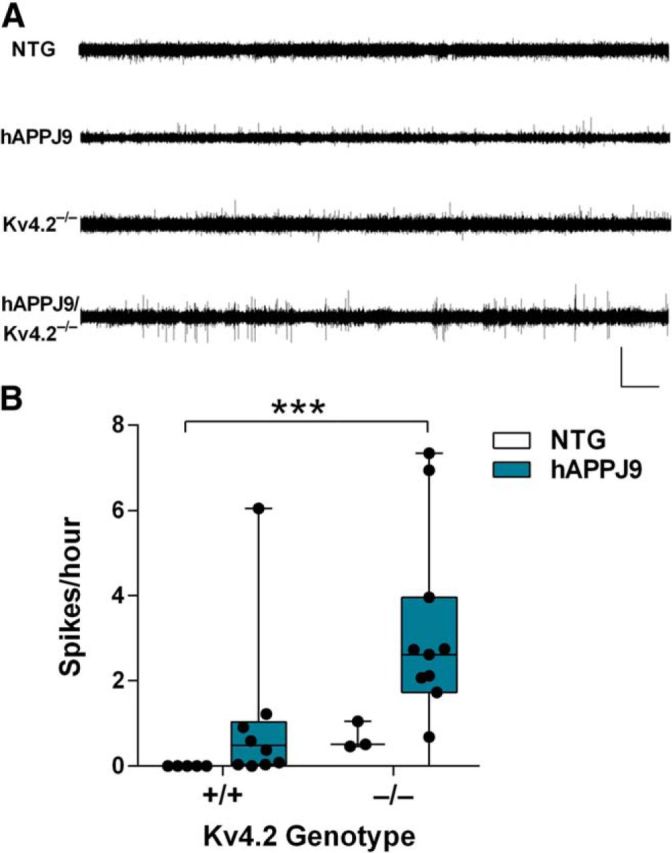
Neuronal hyperexcitability occurs early in the pathogenesis of Alzheimer's disease (AD) and contributes to network dysfunction in AD patients. In other disorders with neuronal hyperexcitability, dysfunction in the dendrites often contributes, but dendritic excitability has not been directly examined in AD models. We used dendritic patch-clamp recordings to measure dendritic excitability in the CA1 region of the hippocampus. We found that dendrites, more so than somata, of hippocampal neurons were hyperexcitable in mice overexpressing Aβ. This dendritic hyperexcitability was associated with depletion of Kv4.2, a dendritic potassium channel important for regulating dendritic excitability and synaptic plasticity. The antiepileptic drug, levetiracetam, blocked Kv4.2 depletion. Tau was required, as crossing with tau knock-out mice also prevented both Kv4.2 depletion and dendritic hyperexcitability. Dendritic hyperexcitability induced by Kv4.2 deficiency exacerbated behavioral deficits and increased epileptiform activity in hAPP mice. We conclude that increased dendritic excitability, associated with changes in dendritic ion channels including Kv4.2, may contribute to neuronal dysfunction in early stages AD.
Keywords: Alzheimer; Kv4.2; amyloid-beta; dendrites; excitability; tau.
Copyright © 2015 the authors 0270-6474/15/356221-10$15.00/0.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous