

Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery?
- PMID: 25878363
- PMCID: PMC4400837
- DOI: 10.1158/1078-0432.CCR-14-3214
Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery?
Abstract
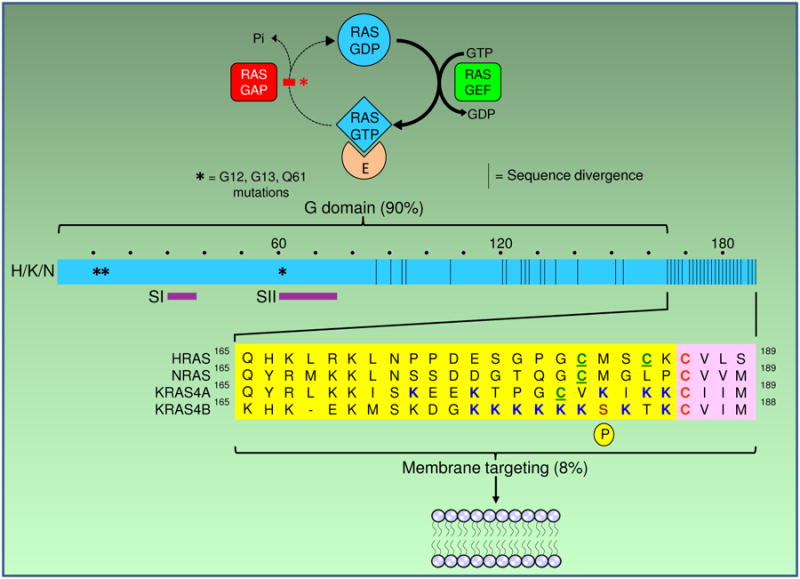
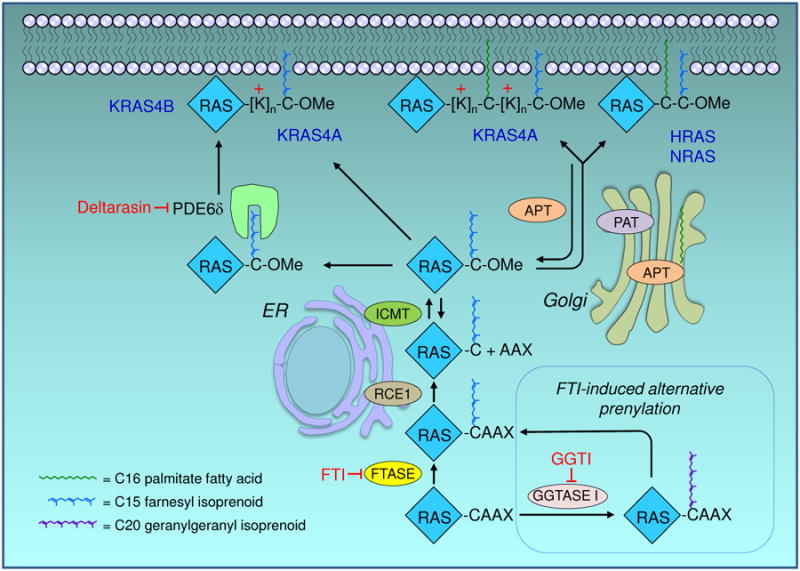
RAS proteins require membrane association for their biologic activity, making this association a logical target for anti-RAS therapeutics. Lipid modification of RAS proteins by a farnesyl isoprenoid is an obligate step in that association, and is an enzymatic process. Accordingly, farnesyltransferase inhibitors (FTI) were developed as potential anti-RAS drugs. The lack of efficacy of FTIs as anticancer drugs was widely seen as indicating that blocking RAS membrane association was a flawed approach to cancer treatment. However, a deeper understanding of RAS modification and trafficking has revealed that this was an erroneous conclusion. In the presence of FTIs, KRAS and NRAS, which are the RAS isoforms most frequently mutated in cancer, become substrates for alternative modification, can still associate with membranes, and can still function. Thus, FTIs failed not because blocking RAS membrane association is an ineffective approach, but because FTIs failed to accomplish that task. Recent findings regarding RAS isoform trafficking and the regulation of RAS subcellular localization have rekindled interest in efforts to target these processes. In particular, improved understanding of the palmitoylation/depalmitoylation cycle that regulates RAS interaction with the plasma membrane, endomembranes, and cytosol, and of the potential importance of RAS chaperones, have led to new approaches. Efforts to validate and target other enzymatically regulated posttranslational modifications are also ongoing. In this review, we revisit lessons learned, describe the current state of the art, and highlight challenging but promising directions to achieve the goal of disrupting RAS membrane association and subcellular localization for anti-RAS drug development. Clin Cancer Res; 21(8); 1819-27. ©2015 AACR. See all articles in this CCR Focus section, "Targeting RAS-Driven Cancers."
©2015 American Association for Cancer Research.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
