

De novo transcriptome assembly of the wild relative of tea tree (Camellia taliensis) and comparative analysis with tea transcriptome identified putative genes associated with tea quality and stress response
- PMID: 25881092
- PMCID: PMC4404113
- DOI: 10.1186/s12864-015-1494-4
De novo transcriptome assembly of the wild relative of tea tree (Camellia taliensis) and comparative analysis with tea transcriptome identified putative genes associated with tea quality and stress response
Abstract
Background: Camellia taliensis is one of the most important wild relatives of cultivated tea tree, C. sinensis. The species extensively occupies mountainous habitats representing a wide-range abiotic tolerance and biotic resistance and thus harbors valuable gene resources that may greatly benefit genetic improvement of cultivated tea tree. However, owning to a large genome size of ~3 Gb and structurally complex genome, there are fairly limited genetic information and particularly few genomic resources publicly available for this species. To better understand the key pathways determining tea flavor and enhance tea tree breeding programs, we performed a high-throughput transcriptome sequencing for C. taliensis.
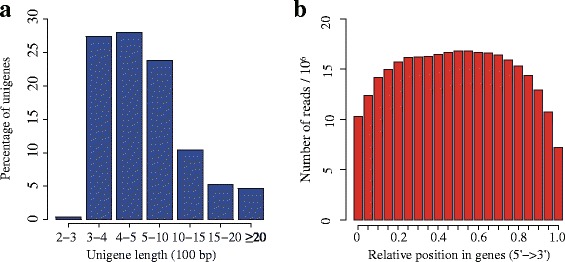
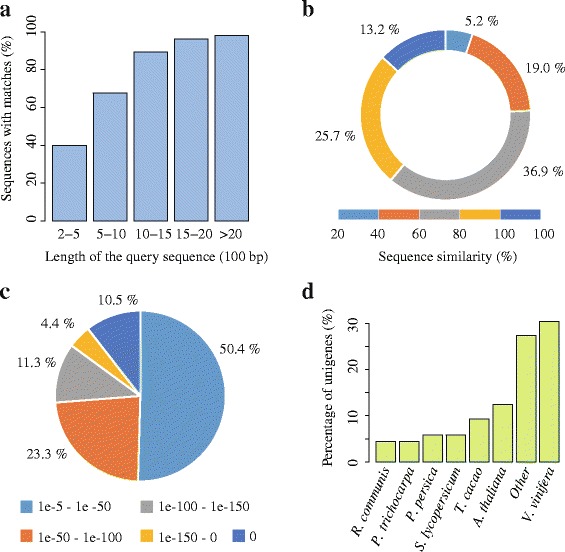
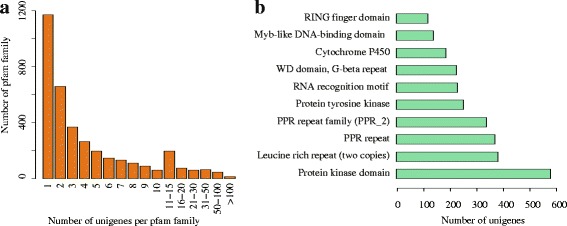
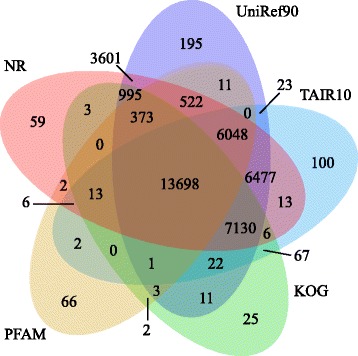
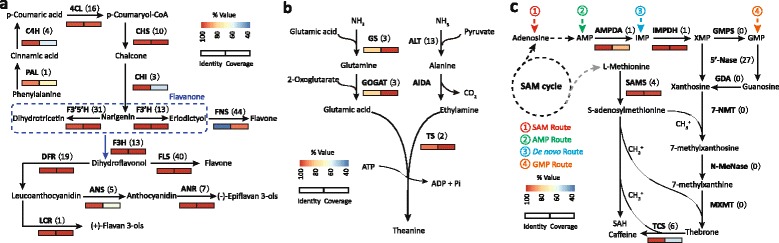
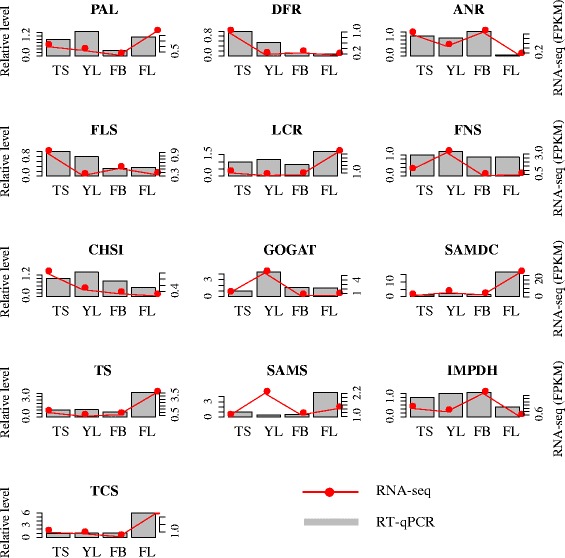
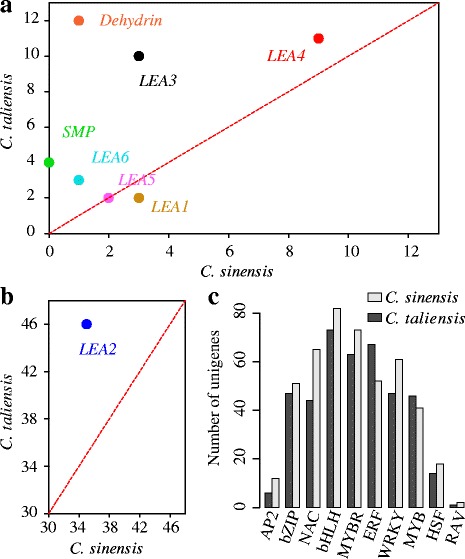
Results: In this study, approximate 241.5 million high-quality paired-end reads, accounting for ~24 Gb of sequence data, were generated from tender shoots, young leaves, flower buds and flowers using Illumina HiSeq 2000 platform. De novo assembly with further processing and filtering yielded a set of 67,923 transcripts with an average length of 685 bp and an N50 of 995 bp. Based on sequence similarity searches against public databases, a total of 39,475 transcripts were annotated with gene descriptions, conserved protein domains or gene ontology (GO) terms. Candidate genes for major metabolic pathways involved in tea quality were identified and experimentally validated using RT-qPCR. Further gene expression profiles showed that they are differentially regulated at different developmental stages. To gain insights into the evolution of these genes, we aligned them to the previously cloned orthologous genes in C. sinensis, and found that considerable nucleotide variation within several genes involved in important secondary metabolic biosynthesis pathways, of which flavone synthase II gene (FNSII) is the most variable between these two species. Moreover, comparative analyses revealed that C. taliensis shows a remarkable expansion of LEA genes, compared to C. sinensis, which might contribute to the observed stronger stress resistance of C. taliensis.
Conclusion: We reported the first large-coverage transcriptome datasets for C. taliensis using the next-generation sequencing technology. Such comprehensive EST datasets provide an unprecedented opportunity for identifying genes involved in several major metabolic pathways and will accelerate functional genomic studies and genetic improvement efforts of tea trees in the future.
Figures

References
-
- Ming TL. A revision of Camellia Section Thea. Acta Botanica Yunnanica. 1992;14(2):116–32.
-
- Takeda Y. Cross compatibility of tea (Camellia sinensis) and its allied species in the genus Camellia. Jpn Agri Res Quar. 1990;24(14):111–6.
-
- Zhang J, Wang PS, Chen HW, Yi B. Wild tea populations in Shuangjiang Mengku of Yunnan Province. J Tea. 2003;29(4):220–1.
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
