

Integrative analysis of survival-associated gene sets in breast cancer
- PMID: 25881247
- PMCID: PMC4359519
- DOI: 10.1186/s12920-015-0086-0
Integrative analysis of survival-associated gene sets in breast cancer
Abstract
Background: Patient gene expression information has recently become a clinical feature used to evaluate breast cancer prognosis. The emergence of prognostic gene sets that take advantage of these data has led to a rich library of information that can be used to characterize the molecular nature of a patient's cancer. Identifying robust gene sets that are consistently predictive of a patient's clinical outcome has become one of the main challenges in the field.
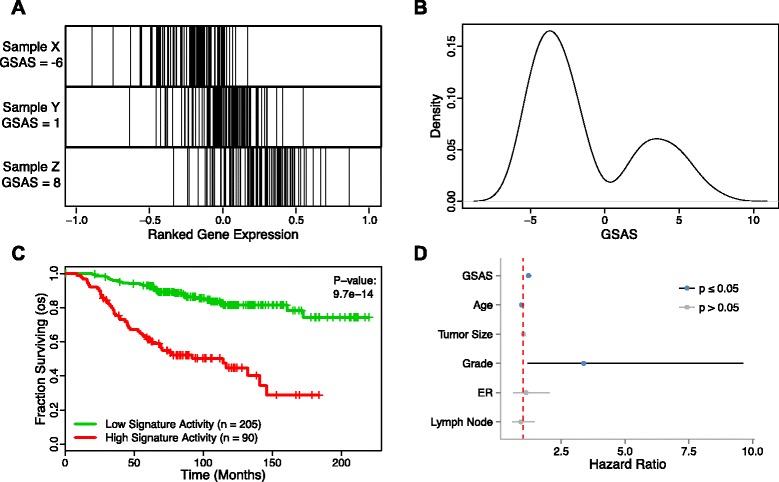
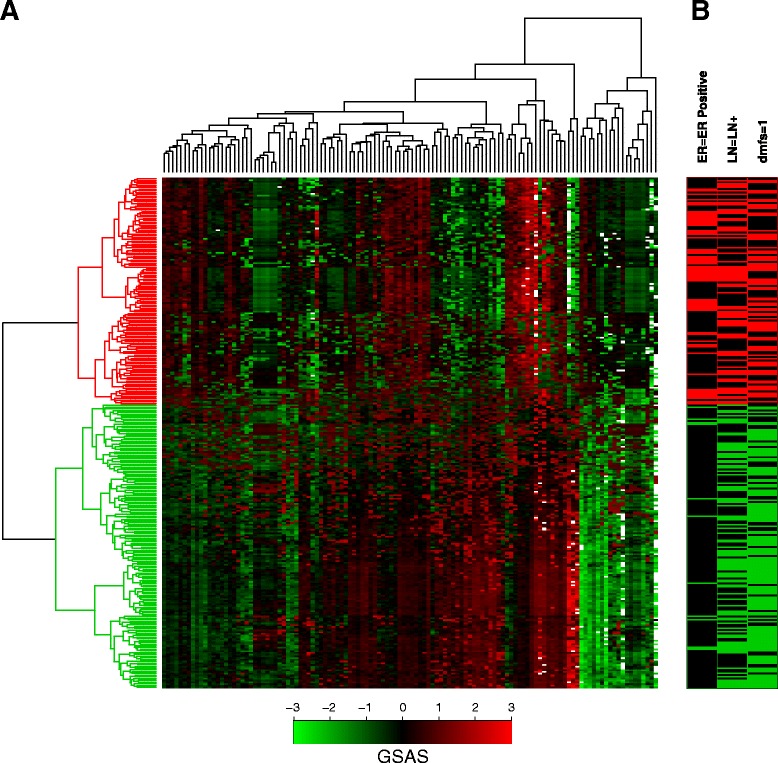
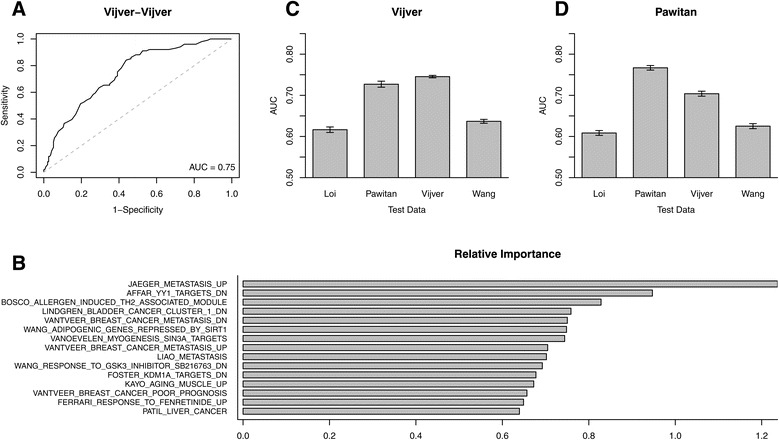
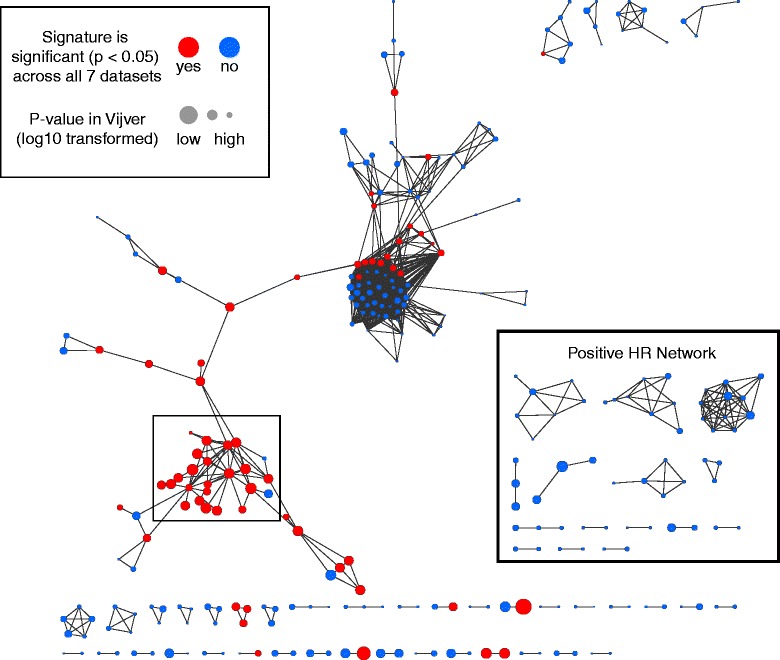
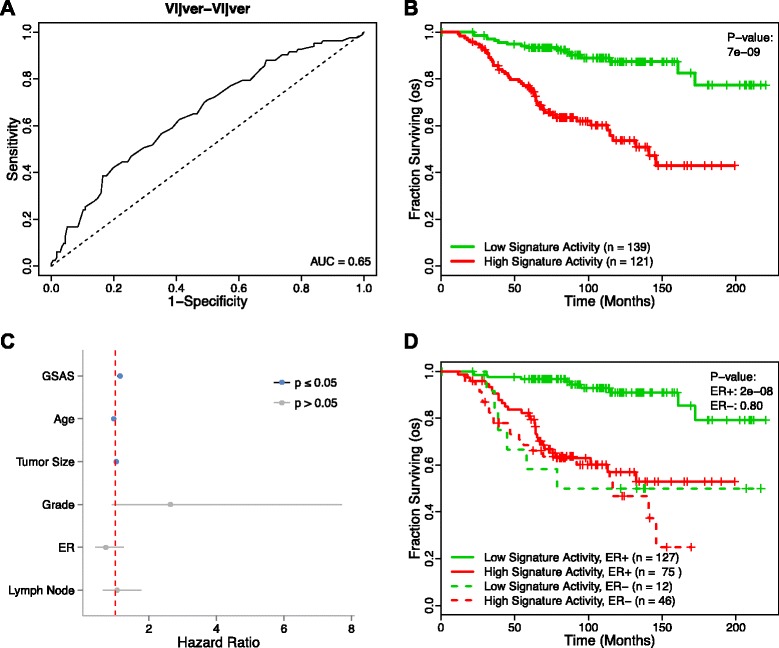
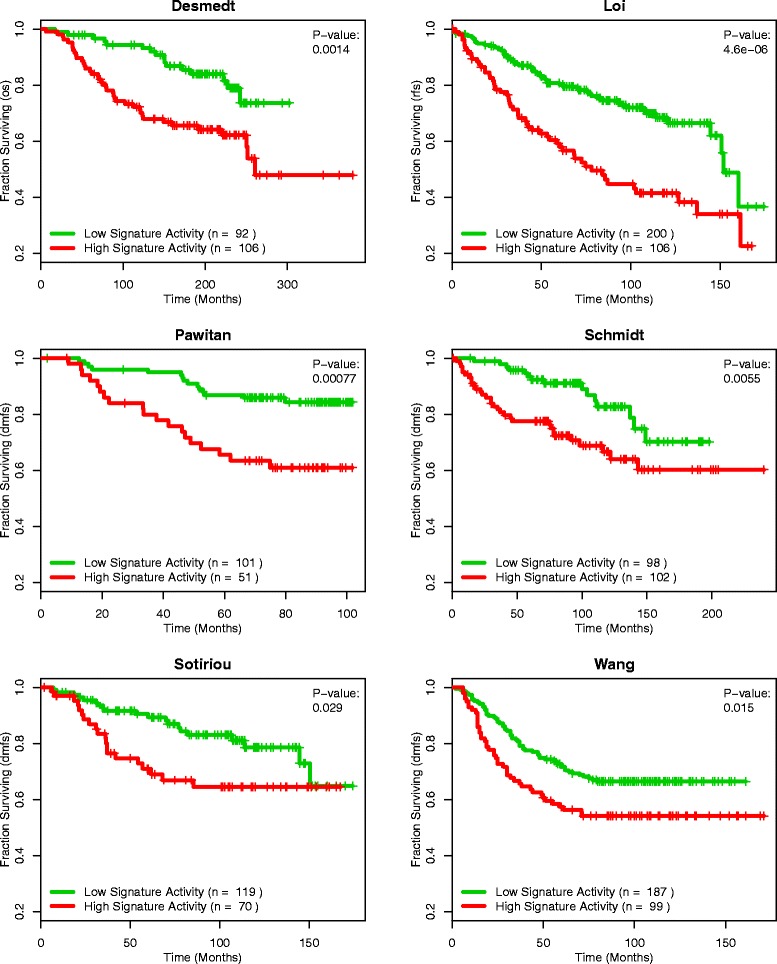
Methods: We inputted our previously established BASE algorithm with patient gene expression data and gene sets from MSigDB to develop the gene set activity score (GSAS), a metric that quantitatively assesses a gene set's activity level in a given patient. We utilized this metric, along with patient time-to-event data, to perform survival analyses to identify the gene sets that were significantly correlated with patient survival. We then performed cross-dataset analyses to identify robust prognostic gene sets and to classify patients by metastasis status. Additionally, we created a gene set network based on component gene overlap to explore the relationship between gene sets derived from MSigDB. We developed a novel gene set based on this network's topology and applied the GSAS metric to characterize its role in patient survival.
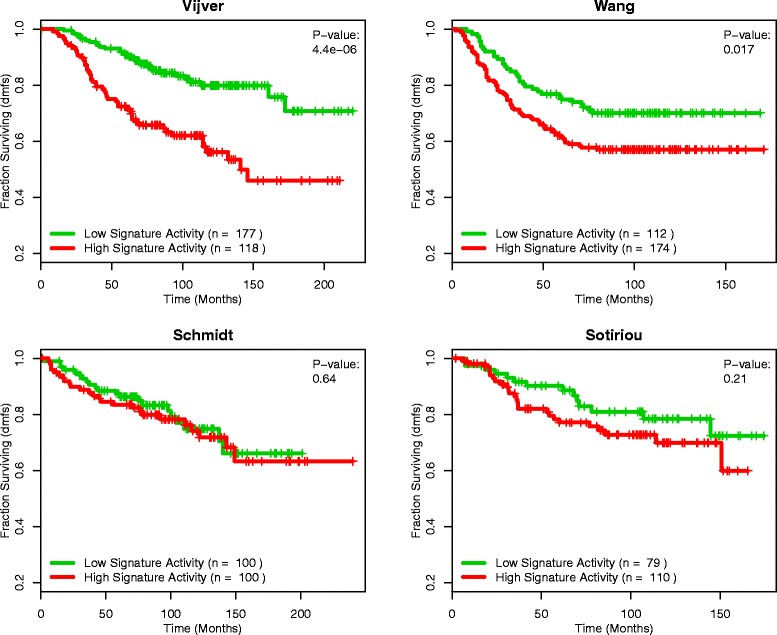
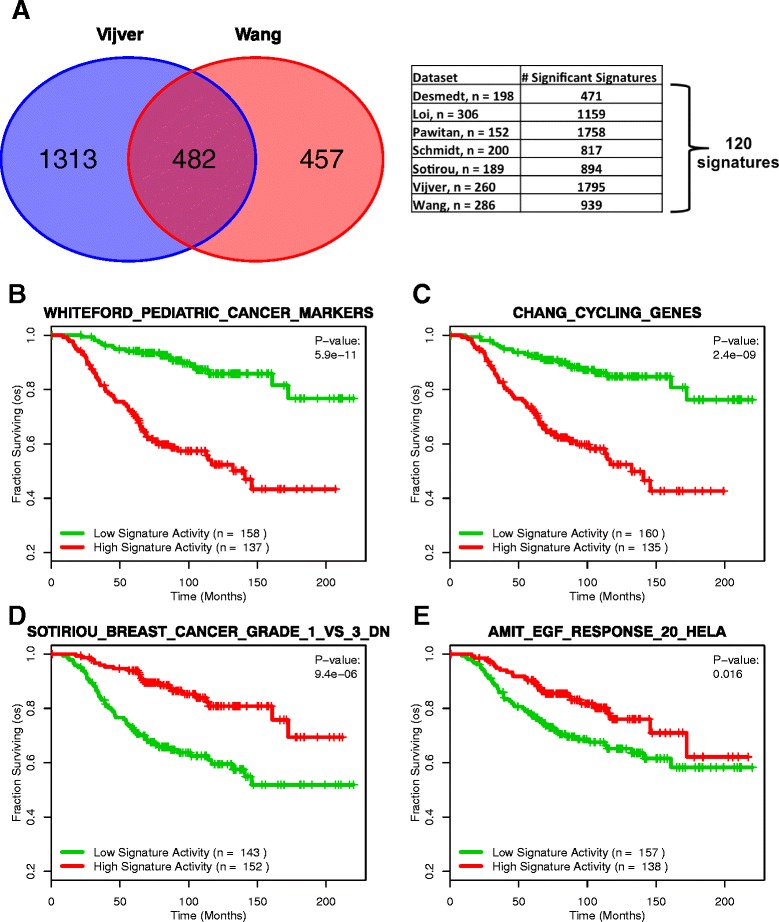
Results: Using the GSAS metric, we identified 120 gene sets that were significantly associated with patient survival in all datasets tested. The gene overlap network analysis yielded a novel gene set enriched in genes shared by the robustly predictive gene sets. This gene set was highly correlated to patient survival when used alone. Most interestingly, removal of the genes in this gene set from the gene pool on MSigDB resulted in a large reduction in the number of predictive gene sets, suggesting a prominent role for these genes in breast cancer progression.
Conclusions: The GSAS metric provided a useful medium by which we systematically investigated how gene sets from MSigDB relate to breast cancer patient survival. We used this metric to identify predictive gene sets and to construct a novel gene set containing genes heavily involved in cancer progression.
Figures
References
-
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101:9309–14. doi: 10.1073/pnas.0401994101. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
