

An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data
- PMID: 25883319
- PMCID: PMC4448687
- DOI: 10.1101/gr.176552.114
An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data
Abstract
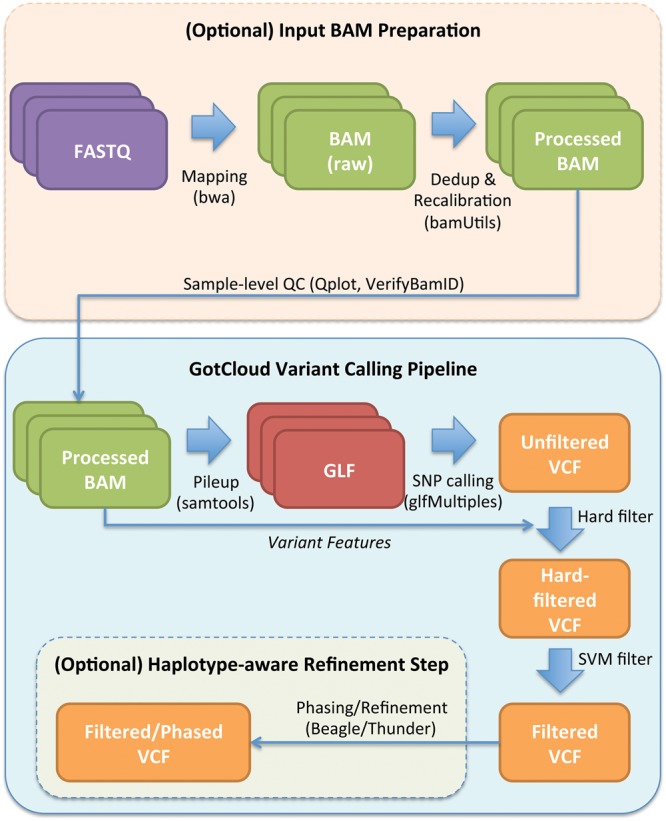
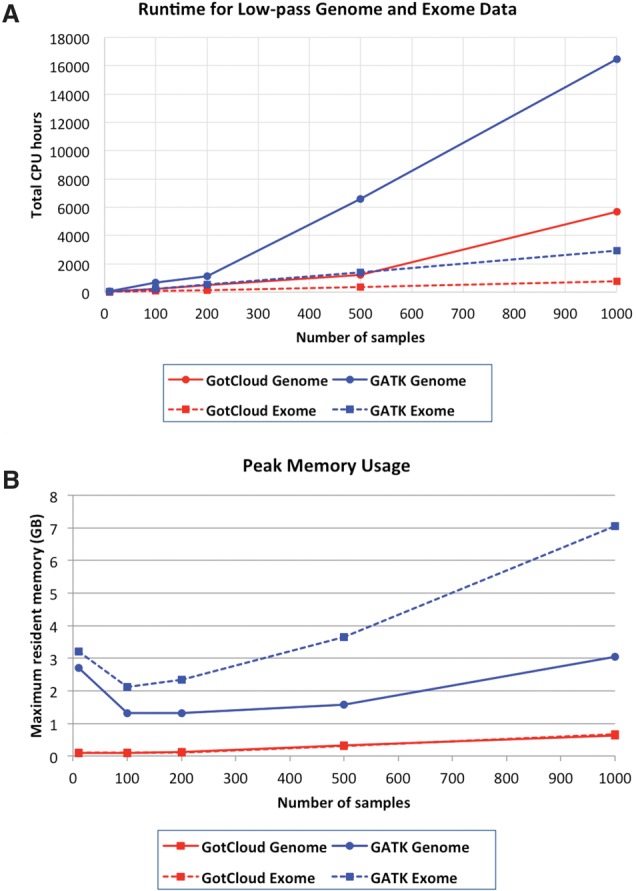
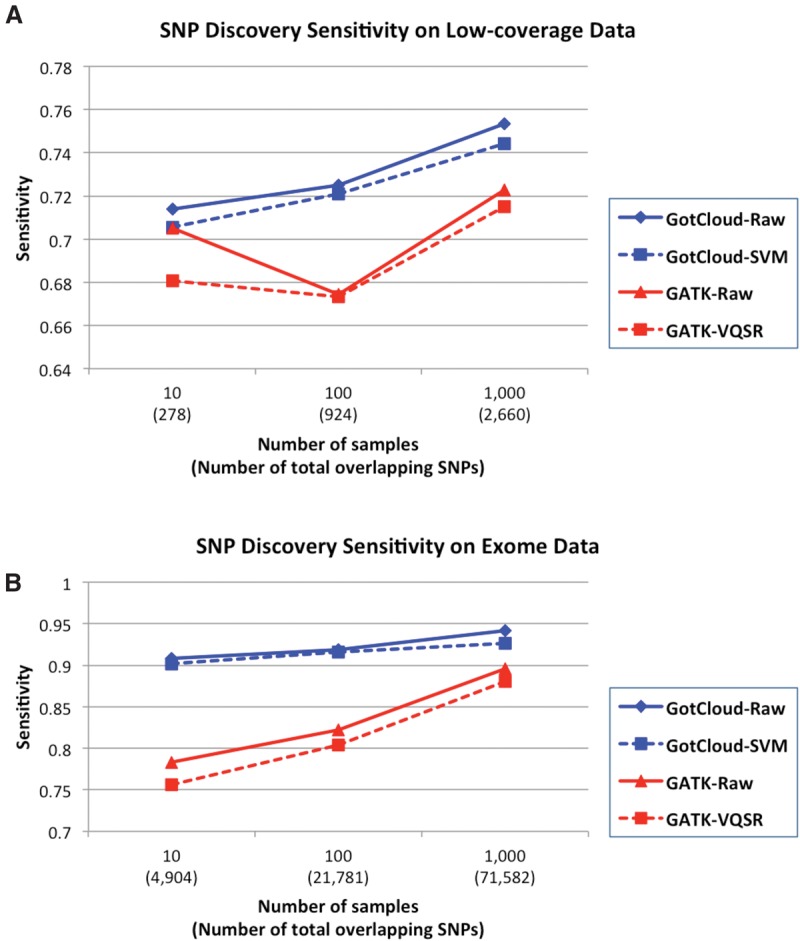
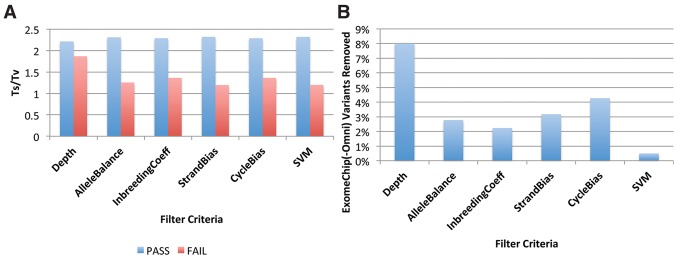
The analysis of next-generation sequencing data is computationally and statistically challenging because of the massive volume of data and imperfect data quality. We present GotCloud, a pipeline for efficiently detecting and genotyping high-quality variants from large-scale sequencing data. GotCloud automates sequence alignment, sample-level quality control, variant calling, filtering of likely artifacts using machine-learning techniques, and genotype refinement using haplotype information. The pipeline can process thousands of samples in parallel and requires less computational resources than current alternatives. Experiments with whole-genome and exome-targeted sequence data generated by the 1000 Genomes Project show that the pipeline provides effective filtering against false positive variants and high power to detect true variants. Our pipeline has already contributed to variant detection and genotyping in several large-scale sequencing projects, including the 1000 Genomes Project and the NHLBI Exome Sequencing Project. We hope it will now prove useful to many medical sequencing studies.
© 2015 Jun et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
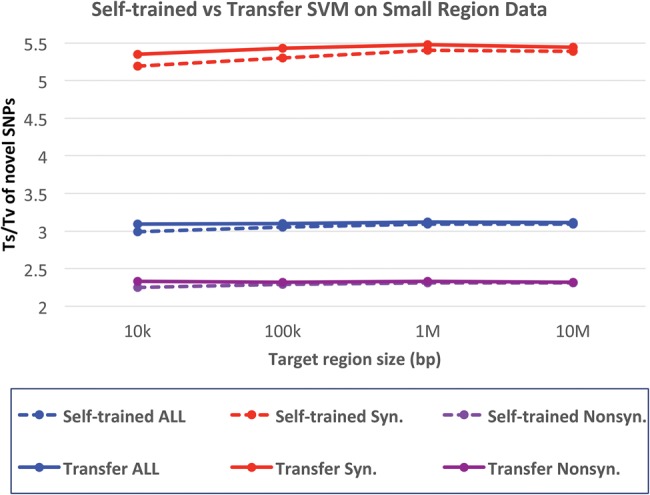
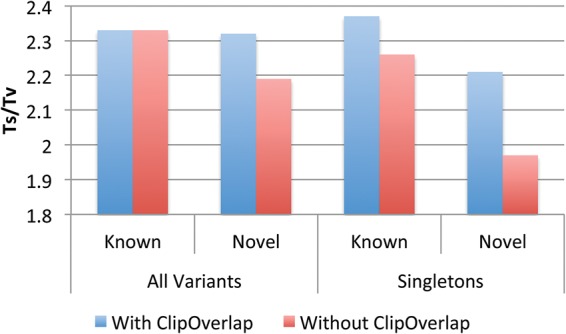
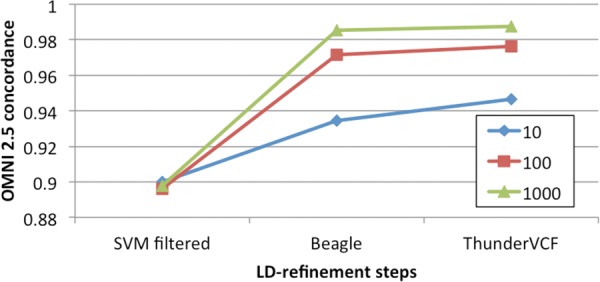
References
-
- Amari S, Wu S. 1999. Improving support vector machine classifiers by modifying kernel functions. Neural Netw 12: 783–789. - PubMed
-
- Chang CC, Lin CJ. 2011. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol 2: 27:1–27:27.
-
- Cortes C, Vapnik V. 1995. Support-vector networks. Mach Learn 20: 273–297.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases