

Rationale, design and objectives of ARegPKD, a European ARPKD registry study
- PMID: 25886171
- PMCID: PMC4359504
- DOI: 10.1186/s12882-015-0002-z
Rationale, design and objectives of ARegPKD, a European ARPKD registry study
Abstract
Background: Autosomal recessive polycystic kidney disease (ARPKD) is a rare but frequently severe disorder that is typically characterized by cystic kidneys and congenital hepatic fibrosis but displays pronounced phenotypic heterogeneity. ARPKD is among the most important causes for pediatric end stage renal disease and a leading reason for liver-, kidney- or combined liver kidney transplantation in childhood. The underlying pathophysiology, the mechanisms resulting in the observed clinical heterogeneity and the long-term clinical evolution of patients remain poorly understood. Current treatment approaches continue to be largely symptomatic and opinion-based even in most-advanced medical centers. While large clinical trials for the frequent and mostly adult onset autosomal dominant polycystic kidney diseases have recently been conducted, therapeutic initiatives for ARPKD are facing the challenge of small and clinically variable cohorts for which reliable end points are hard to establish.
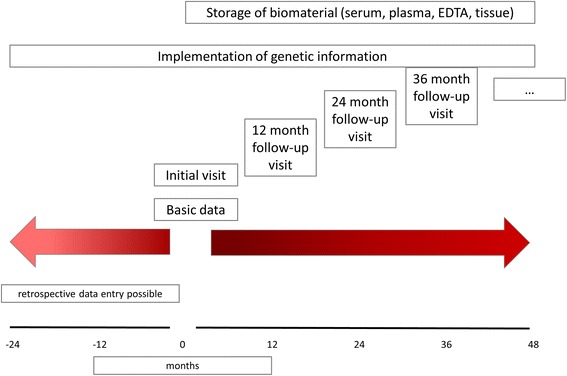
Methods/design: ARegPKD is an international, mostly European, observational study to deeply phenotype ARPKD patients in a pro- and retrospective fashion. This registry study is conducted with the support of the German Society for Pediatric Nephrology (GPN) and the European Study Consortium for Chronic Kidney Disorders Affecting Pediatric Patients (ESCAPE Network). ARegPKD clinically characterizes long-term ARPKD courses by a web-based approach that uses detailed basic data questionnaires in combination with yearly follow-up visits. Clinical data collection is accompanied by associated biobanking and reference histology, thus setting roots for future translational research.
Discussion: The novel registry study ARegPKD aims to characterize miscellaneous subcohorts and to compare the applied treatment options in a large cohort of deeply characterized patients. ARegPKD will thus provide evidence base for clinical treatment decisions and contribute to the pathophysiological understanding of this severe inherited disorder.
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
