

Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria × ananassa
- PMID: 25886969
- PMCID: PMC4374422
- DOI: 10.1186/s12864-015-1310-1
Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria × ananassa
Abstract
Background: A high-throughput genotyping platform is needed to enable marker-assisted breeding in the allo-octoploid cultivated strawberry Fragaria × ananassa. Short-read sequences from one diploid and 19 octoploid accessions were aligned to the diploid Fragaria vesca 'Hawaii 4' reference genome to identify single nucleotide polymorphisms (SNPs) and indels for incorporation into a 90 K Affymetrix® Axiom® array. We report the development and preliminary evaluation of this array.
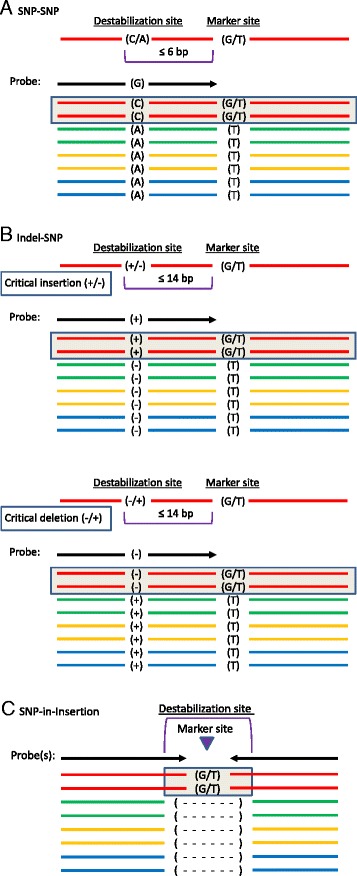
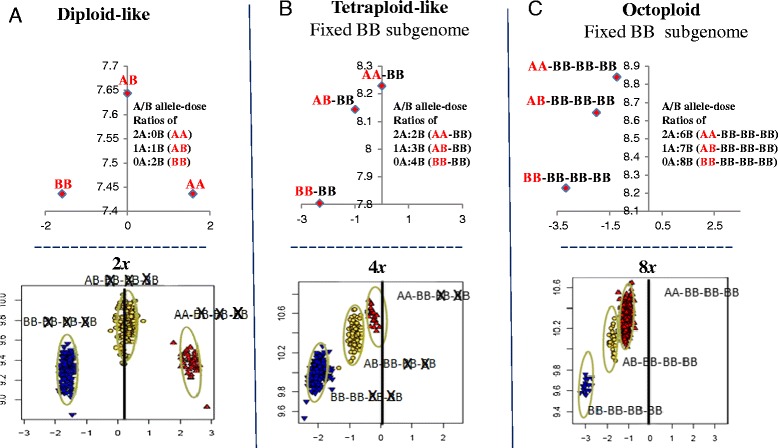
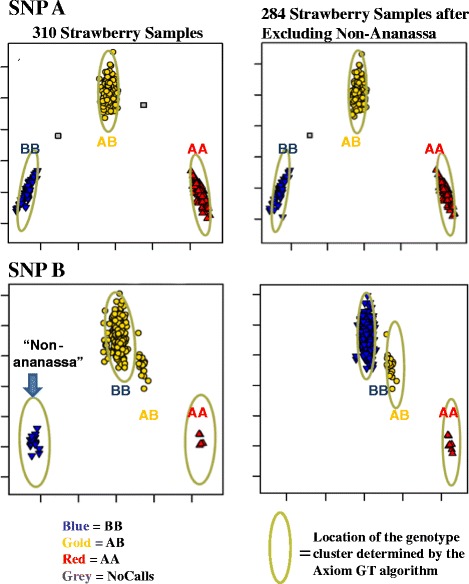
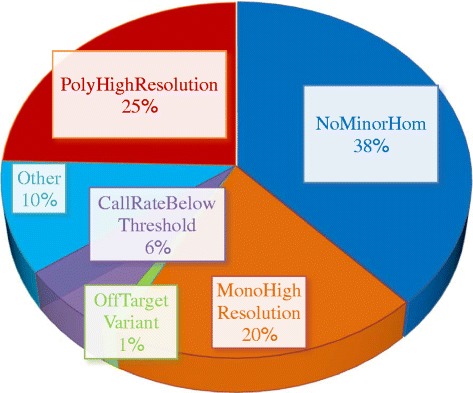
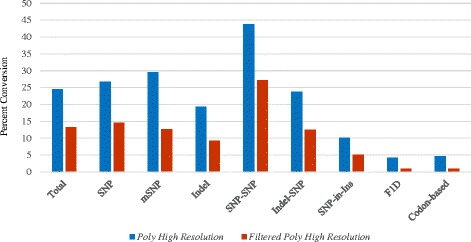
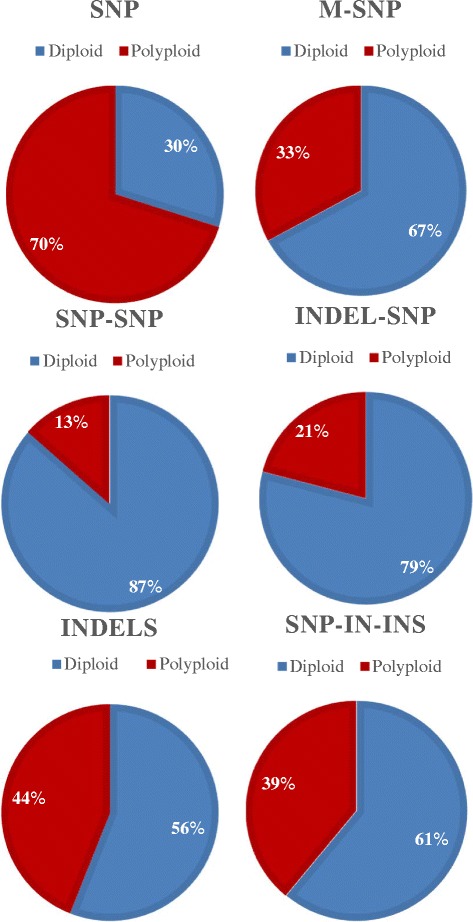
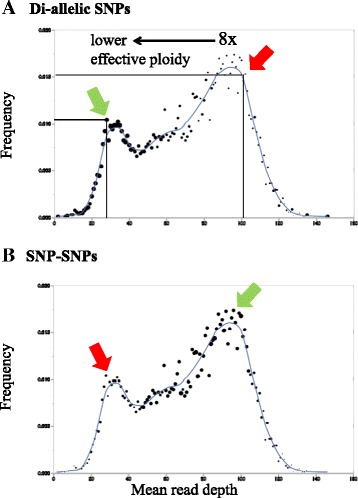
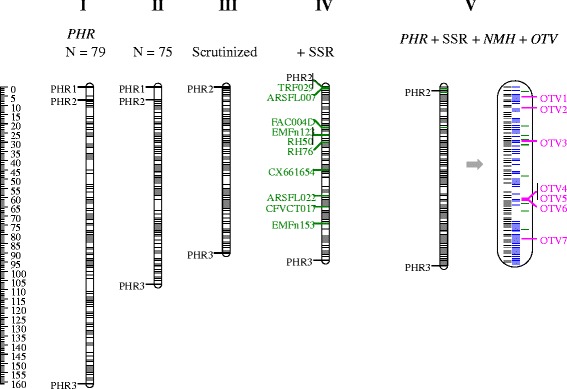
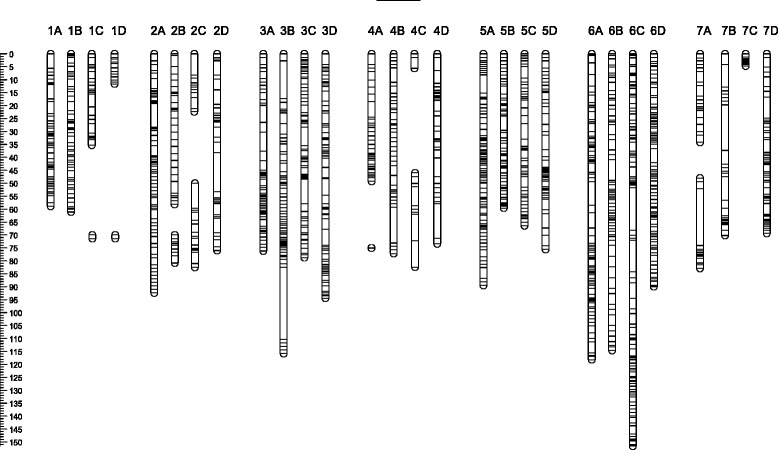
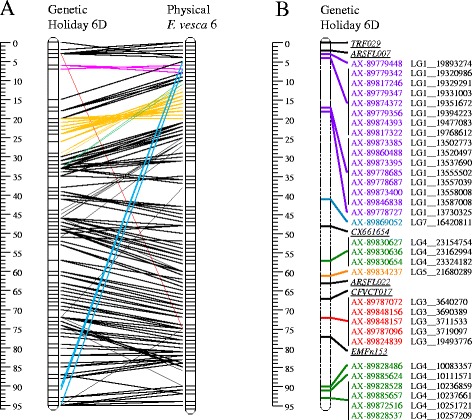
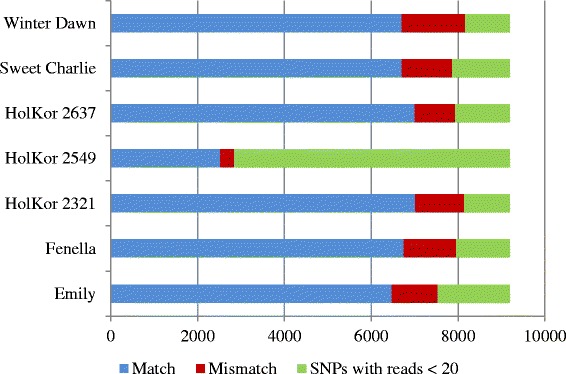
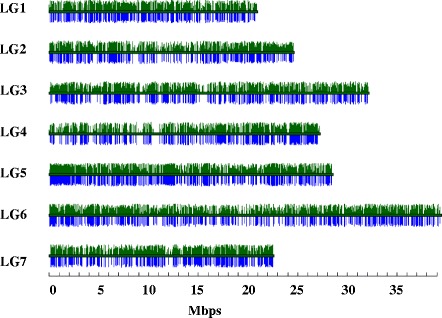
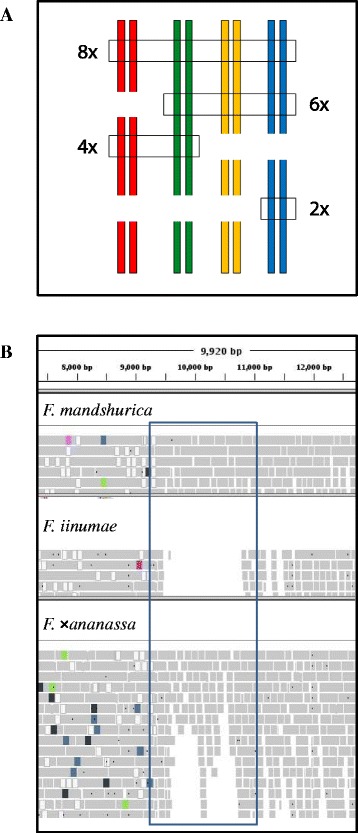
Results: About 36 million sequence variants were identified in a 19 member, octoploid germplasm panel. Strategies and filtering pipelines were developed to identify and incorporate markers of several types: di-allelic SNPs (66.6%), multi-allelic SNPs (1.8%), indels (10.1%), and ploidy-reducing "haploSNPs" (11.7%). The remaining SNPs included those discovered in the diploid progenitor F. iinumae (3.9%), and speculative "codon-based" SNPs (5.9%). In genotyping 306 octoploid accessions, SNPs were assigned to six classes with Affymetrix's "SNPolisher" R package. The highest quality classes, PolyHigh Resolution (PHR), No Minor Homozygote (NMH), and Off-Target Variant (OTV) comprised 25%, 38%, and 1% of array markers, respectively. These markers were suitable for genetic studies as demonstrated in the full-sib family 'Holiday' × 'Korona' with the generation of a genetic linkage map consisting of 6,594 PHR SNPs evenly distributed across 28 chromosomes with an average density of approximately one marker per 0.5 cM, thus exceeding our goal of one marker per cM.
Conclusions: The Affymetrix IStraw90 Axiom array is the first high-throughput genotyping platform for cultivated strawberry and is commercially available to the worldwide scientific community. The array's high success rate is likely driven by the presence of naturally occurring variation in ploidy level within the nominally octoploid genome, and by effectiveness of the employed array design and ploidy-reducing strategies. This array enables genetic analyses including generation of high-density linkage maps, identification of quantitative trait loci for economically important traits, and genome-wide association studies, thus providing a basis for marker-assisted breeding in this high value crop.
Figures


References
-
- Iezzoni A, Weebadde C, Luby J, Chengyan Y, van de Weg E, Fazio G, et al. RosBREED: Enabling marker-assisted breeding in Rosaceae. Acta Horticult. 2010;859:389–94.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
