

Mutations in TTC19: expanding the molecular, clinical and biochemical phenotype
- PMID: 25887401
- PMCID: PMC4422538
- DOI: 10.1186/s13023-015-0254-5
Mutations in TTC19: expanding the molecular, clinical and biochemical phenotype
Abstract
Background: TTC19 deficiency is a progressive neurodegenerative disease associated with isolated mitochondrial respiratory chain (MRC) complex III deficiency and loss-of-function mutations in the TT19 gene in the few patients reported so far.
Methods: We performed exome sequencing and selective mutational analysis of TTC19, respectively, in patients from three unrelated families presenting with initially unspecific clinical signs of muscular hypotonia and global developmental delay followed by regression, ataxia, loss of speech, and rapid neurological deterioration. One patient showed severe lactic acidosis at the neonatal age and during intercurrent illness.
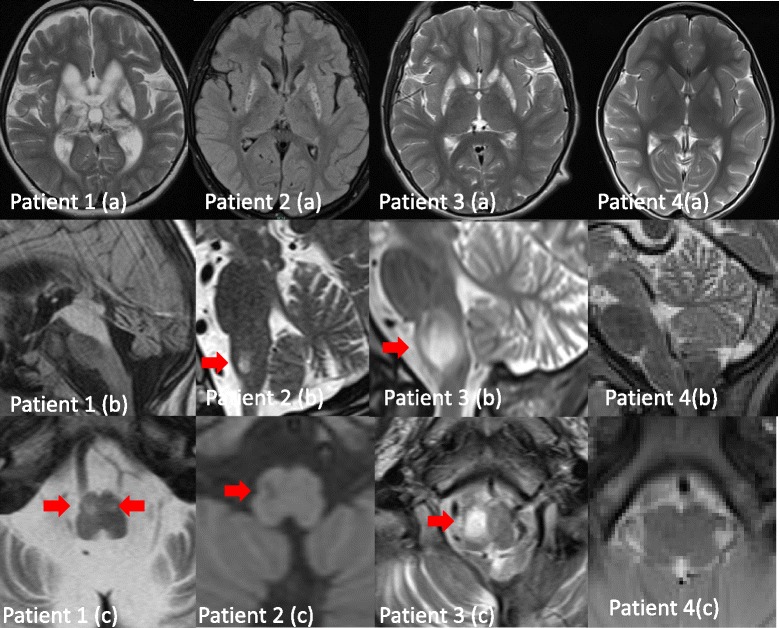
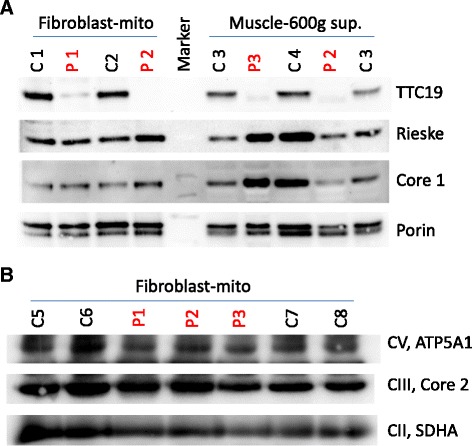
Results: We identified homozygous mutations in all three index cases, in two families novel missense mutations (c.544 T > C/p.Leu185Pro; c.917 T > C/p.Leu324Pro). The younger sister of the severely affected patient 3 showed only mild delay of motor skills and muscular hypotonia so far but is also homozygous for the same mutation. Notably, one patient revealed normal activities of MRC complex III in two independent muscle biopsies. Neuroimaging of the severely affected patients demonstrated lesions in putamen and caudate nuclei, cerebellar atrophy, and the unusual finding of hypertrophic olivary nuclei degeneration. Reviewing the literature revealed striking similarities regarding neuroimaging and clinical course in pediatric patients with TTC19 deficiency: patterns consistent with Leigh or Leigh-like syndrome were found in almost all, hypertrophic olivary nucleus degeneration in all patients reported so far. The clinical course in pediatric patients is characterized by an initially unspecific developmental delay, followed by regression, progressive signs and symptoms of cerebellar, basal ganglia and brainstem affection, especially loss of speech and ataxia. Subsequently, neurological deterioration leading to a vegetative state occurs.
Conclusions: Our findings add to the phenotypic, genetic, and biochemical spectrum of TTC19 deficiency. However, TTC19 deficient patients do show characteristic clinical and neuroimaging features, which may facilitate diagnosis of this yet rare disorder. Normal MRC complex III activity does not exclude the diagnosis.
Figures


References
-
- Kunii M, Doi H, Higashiyama Y, Kugimoto C, Ueda N, Hirata J, et al. A Japanese case of cerebellar ataxia, spastic paraparesis and deep sensory impairment associated with a novel homozygous TTC19 mutation. J Human Genet. 2015;14:43–5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
