

Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: a retrospective observational study
- PMID: 25887606
- PMCID: PMC4407793
- DOI: 10.1186/s13023-015-0259-0
Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: a retrospective observational study
Abstract
Background: Mucopolysaccharidosis II (MPS II) is associated with a broad spectrum of chronic and progressive, life-limiting symptoms. Idursulfase is approved for MPS II enzyme replacement therapy (ERT) in over 50 countries. This retrospective study evaluated the MPS II burden, organization of clinical care, and effects of idursulfase treatment on the disease in France.
Methods: MPS II patients who had received idursulfase ERT in the French healthcare system were enrolled. In addition to clinician and patient questionnaires, the Clinical Global Impression-Improvement (CGI-I); Patient Global Impression-Improvement (PGI-I); KIDSCREEN-27, and EuroQoL-5D for adult patients scales were used to assess quality of life (QoL) and efficacy.
Results: Fifty-two patients were enrolled from 5 sites in France. The majority of patients (69.2%) presented a severe MPS II phenotype with progressive neurocognitive impairment. Major impacts on QoL were apparent, with at least 1 member of the family having to reorganize working hours (45.5%) or to stop working (22.7%). KIDSCREEN-27 and EuroQoL-5D scale scores were well below those for referent (control) populations. Most families (70.0%) experienced a diagnostic delay of at least 3 years after the initial observation of symptoms. The MPS II diagnosis was often delivered without adequate sensitivity, psychological support, or comprehensive information about the disease. The study population had received a mean of 3.8 ± 1.3 years ERT. Forty-four percent of patients with the attenuated phenotype (without progressive neurocognitive impairment) showed symptom improvement during both the first year (Period 1) and from the end of the first year of treatment to "the present" (Period 2), as measured by CGI-I/PGI-I. 30.3% and 9.1% of severe patients experienced symptom improvement during Periods 1 and 2, respectively, while 63.6% and 51.5% displayed no change. The most common adverse reactions reported were skin rash and other infusion-associated reactions.
Conclusions: MPS II adversely affects multiple domains of QoL for patients and families, requiring multiple healthcare services and social aid programs. The majority of patients with either phenotype experienced either improvement or stability in their symptoms during the first year of ERT, but this was clearly less so for patients with the severe phenotype after the first year of treatment.
Figures
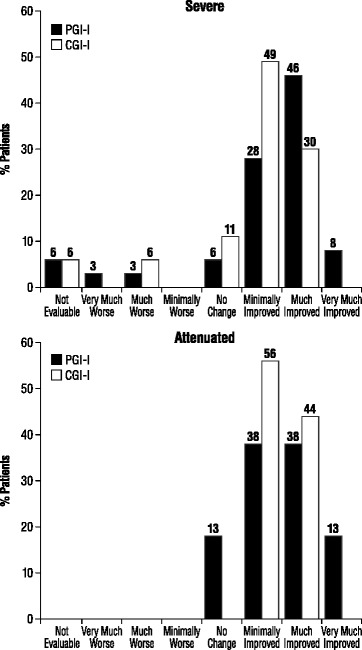
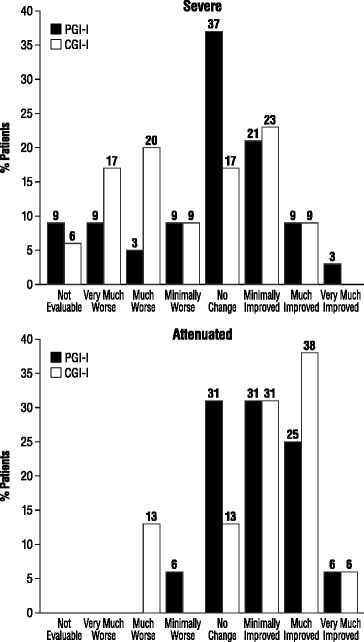
References
-
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. New York: McGraw-Hill; 2001. pp. 3421–52.
-
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–77. doi: 10.1007/s00431-007-0635-4. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
