

Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease
- PMID: 25887922
- PMCID: PMC4414286
- DOI: 10.1186/s13059-015-0637-x
Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease
Abstract
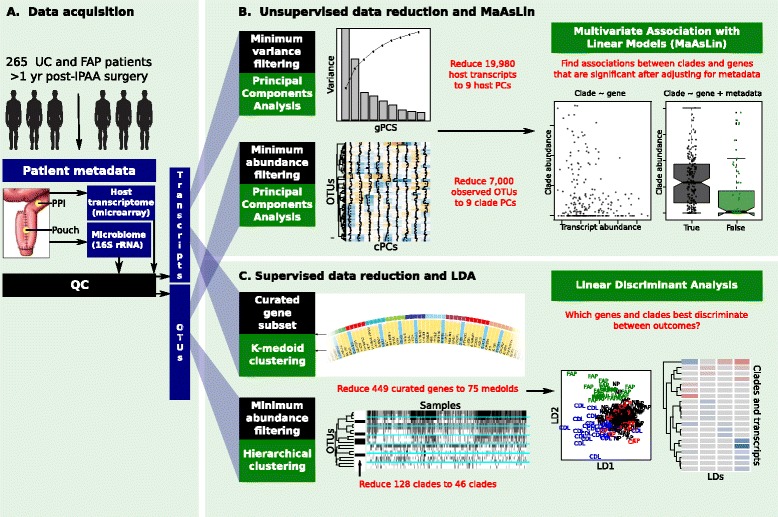
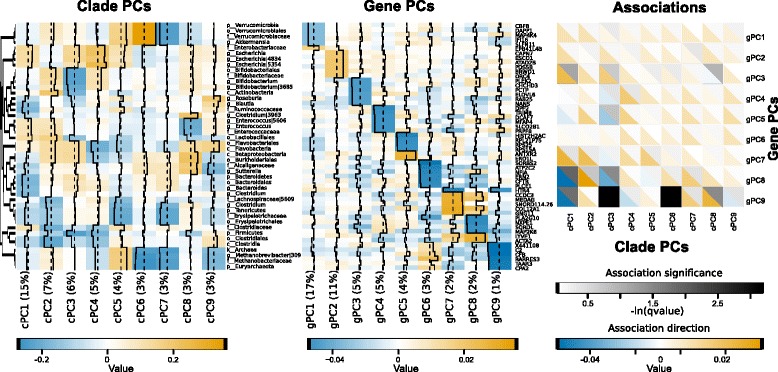
Background: Pouchitis is common after ileal pouch-anal anastomosis (IPAA) surgery for ulcerative colitis (UC). Similar to inflammatory bowel disease (IBD), both host genetics and the microbiota are implicated in its pathogenesis. We use the IPAA model of IBD to associate mucosal host gene expression with mucosal microbiomes and clinical outcomes. We analyze host transcriptomic data and 16S rRNA gene sequencing data from paired biopsies from IPAA patients with UC and familial adenomatous polyposis. To achieve power for a genome-wide microbiome-transcriptome association study, we use principal component analysis for transcript and clade reduction, and identify significant co-variation between clades and transcripts.
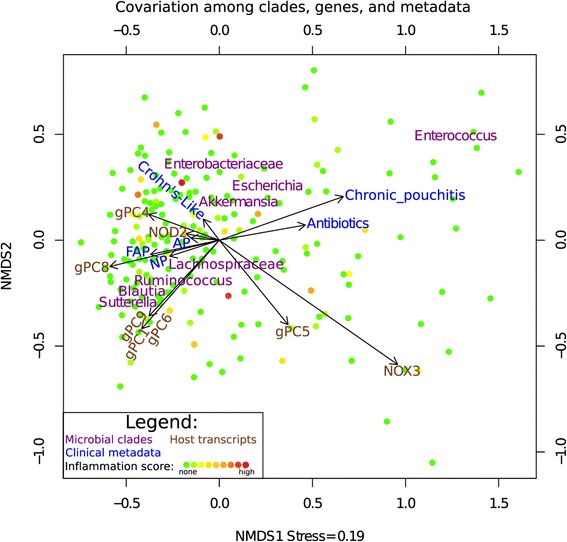
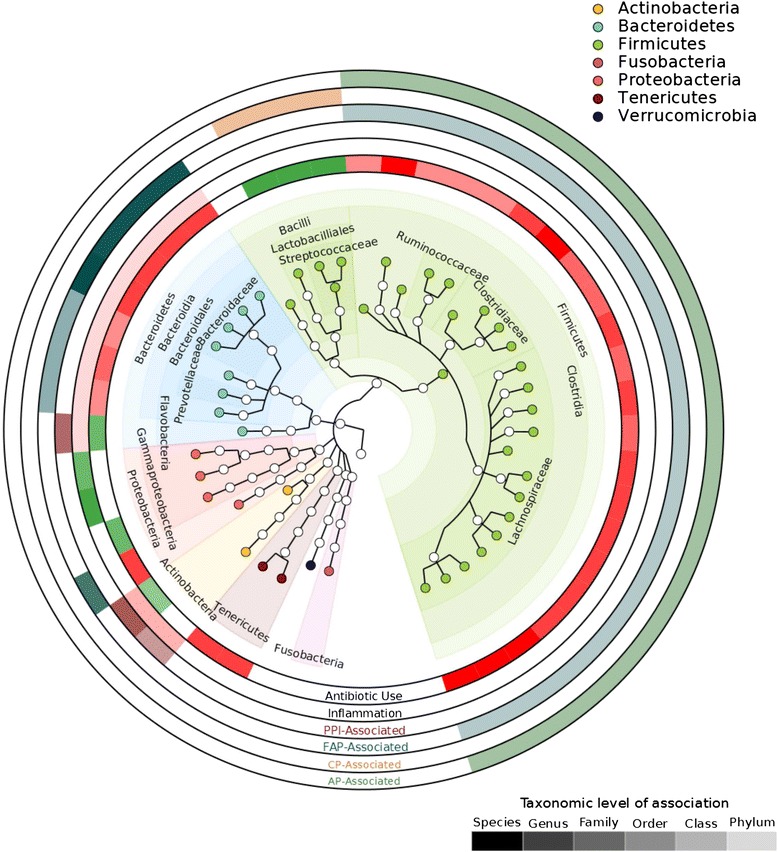
Results: Host transcripts co-vary primarily with biopsy location and inflammation, while microbes co-vary primarily with antibiotic use. Transcript-microbe associations are surprisingly modest, but the most strongly microbially-associated host transcript pattern is enriched for complement cascade genes and for the interleukin-12 pathway. Activation of these host processes is inversely correlated with Sutterella, Akkermansia, Bifidobacteria, and Roseburia abundance, and positively correlated with Escherichia abundance.
Conclusions: This study quantifies the effects of inflammation, antibiotic use, and biopsy location upon the microbiome and host transcriptome during pouchitis. Understanding these effects is essential for basic biological insights as well as for well-designed and adequately-powered studies. Additionally, our study provides a method for profiling host-microbe interactions with appropriate statistical power using high-throughput sequencing, and suggests that cross-sectional changes in gut epithelial transcription are not a major component of the host-microbiome regulatory interface during pouchitis.
Figures
Similar articles
-
Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis.Inflamm Bowel Dis. 2011 May;17(5):1092-100. doi: 10.1002/ibd.21460. Epub 2010 Sep 15. Inflamm Bowel Dis. 2011. PMID: 20845425
-
Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites.Cells. 2021 Nov 19;10(11):3243. doi: 10.3390/cells10113243. Cells. 2021. PMID: 34831464 Free PMC article.
-
Pouch Inflammation Is Associated With a Decrease in Specific Bacterial Taxa.Gastroenterology. 2015 Sep;149(3):718-27. doi: 10.1053/j.gastro.2015.05.041. Epub 2015 May 27. Gastroenterology. 2015. PMID: 26026389
-
Pouchitis: what every gastroenterologist needs to know.Clin Gastroenterol Hepatol. 2013 Dec;11(12):1538-49. doi: 10.1016/j.cgh.2013.03.033. Epub 2013 Apr 16. Clin Gastroenterol Hepatol. 2013. PMID: 23602818 Review.
-
The Microbiome as a Therapy in Pouchitis and Ulcerative Colitis.Nutrients. 2021 May 23;13(6):1780. doi: 10.3390/nu13061780. Nutrients. 2021. PMID: 34071065 Free PMC article. Review.
Cited by
-
Microbiota and mucosal gene expression of fecal microbiota transplantation or placebo treated patients with chronic pouchitis.Gut Microbes. 2024 Jan-Dec;16(1):2295445. doi: 10.1080/19490976.2023.2295445. Epub 2024 Jan 12. Gut Microbes. 2024. PMID: 38214604 Free PMC article.
-
Gut Microbiota Has a Widespread and Modifiable Effect on Host Gene Regulation.mSystems. 2019 Sep 3;4(5):e00323-18. doi: 10.1128/mSystems.00323-18. mSystems. 2019. PMID: 31481602 Free PMC article.
-
Gut Sphingolipid Composition as a Prelude to Necrotizing Enterocolitis.Sci Rep. 2018 Jul 20;8(1):10984. doi: 10.1038/s41598-018-28862-4. Sci Rep. 2018. PMID: 30030452 Free PMC article.
-
LOCOM: A logistic regression model for testing differential abundance in compositional microbiome data with false discovery rate control.Proc Natl Acad Sci U S A. 2022 Jul 26;119(30):e2122788119. doi: 10.1073/pnas.2122788119. Epub 2022 Jul 22. Proc Natl Acad Sci U S A. 2022. PMID: 35867822 Free PMC article.
-
The application of multi-omics in the respiratory microbiome: Progresses, challenges and promises.Comput Struct Biotechnol J. 2023 Oct 12;21:4933-4943. doi: 10.1016/j.csbj.2023.10.016. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 37867968 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
- BioProject/PRJNA269954
- Actions
- Actions
- dbGaP/PHS000659.V1.P1
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
