

aTRAM - automated target restricted assembly method: a fast method for assembling loci across divergent taxa from next-generation sequencing data
- PMID: 25887972
- PMCID: PMC4380108
- DOI: 10.1186/s12859-015-0515-2
aTRAM - automated target restricted assembly method: a fast method for assembling loci across divergent taxa from next-generation sequencing data
Abstract
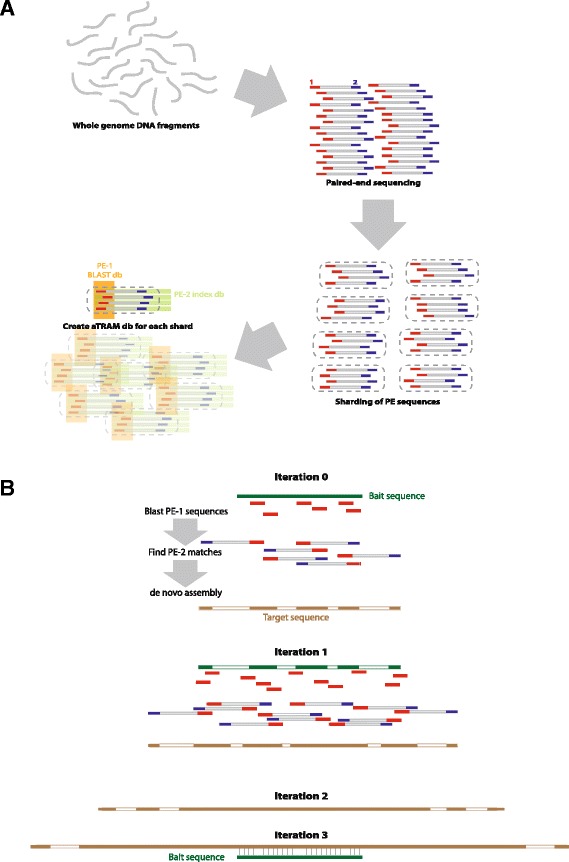
Background: Assembling genes from next-generation sequencing data is not only time consuming but computationally difficult, particularly for taxa without a closely related reference genome. Assembling even a draft genome using de novo approaches can take days, even on a powerful computer, and these assemblies typically require data from a variety of genomic libraries. Here we describe software that will alleviate these issues by rapidly assembling genes from distantly related taxa using a single library of paired-end reads: aTRAM, automated Target Restricted Assembly Method. The aTRAM pipeline uses a reference sequence, BLAST, and an iterative approach to target and locally assemble the genes of interest.
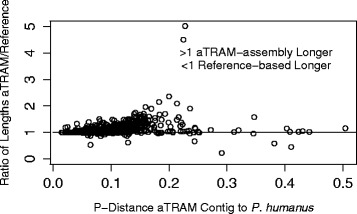
Results: Our results demonstrate that aTRAM rapidly assembles genes across distantly related taxa. In comparative tests with a closely related taxon, aTRAM assembled the same sequence as reference-based and de novo approaches taking on average < 1 min per gene. As a test case with divergent sequences, we assembled >1,000 genes from six taxa ranging from 25 - 110 million years divergent from the reference taxon. The gene recovery was between 97 - 99% from each taxon.
Conclusions: aTRAM can quickly assemble genes across distantly-related taxa, obviating the need for draft genome assembly of all taxa of interest. Because aTRAM uses a targeted approach, loci can be assembled in minutes depending on the size of the target. Our results suggest that this software will be useful in rapidly assembling genes for phylogenomic projects covering a wide taxonomic range, as well as other applications. The software is freely available http://www.github.com/juliema/aTRAM .
Figures
References
-
- Do K, Qin ZS, Vannucci M. 2010. Advances in Statistical Bioinformatics Models and Integrative Inference for High-Throughput Data. Camb Univ Press
-
- Li C, Hofreiter M, Straube N, Corrigan S, Naylor GJP. Capturing protein-coding genes across highly divergent species. Biotechniques. 2013;54:321–6. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
