

Epigenomic evolution in diffuse large B-cell lymphomas
- PMID: 25891015
- PMCID: PMC4411286
- DOI: 10.1038/ncomms7921
Epigenomic evolution in diffuse large B-cell lymphomas
Abstract
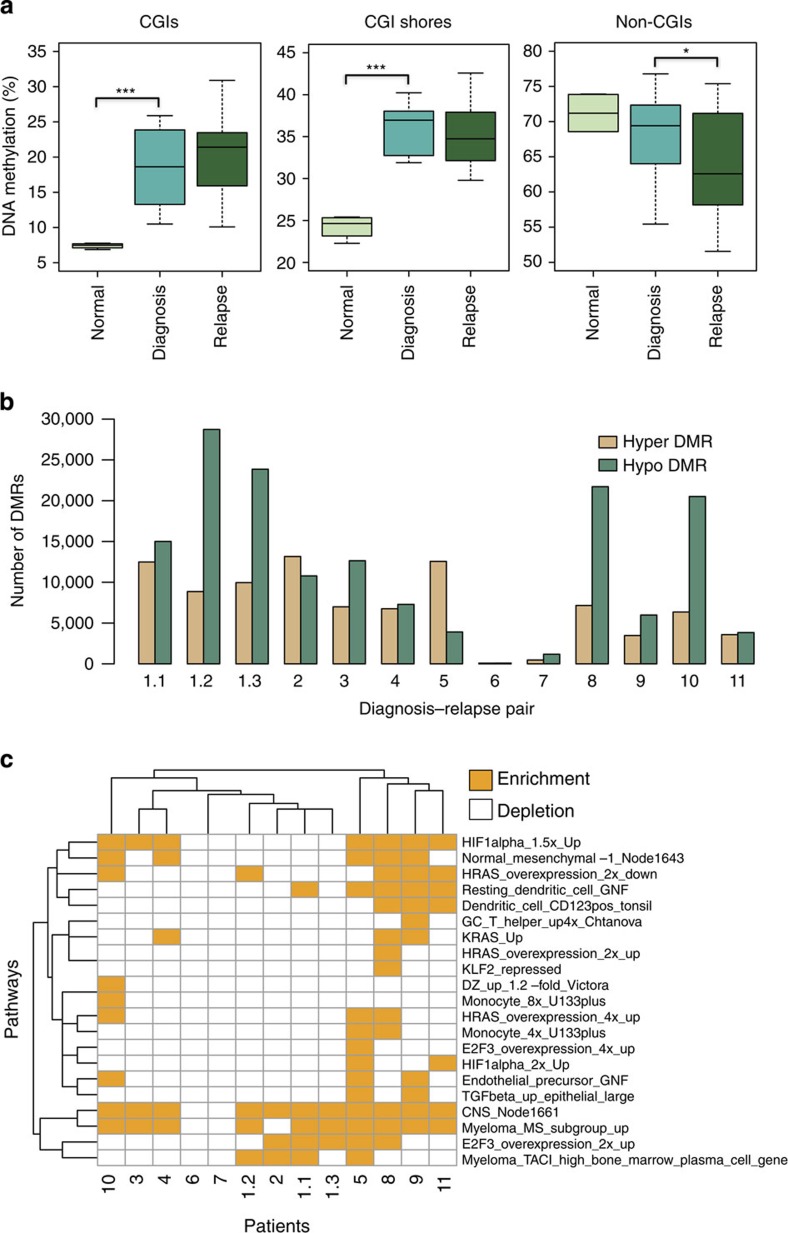
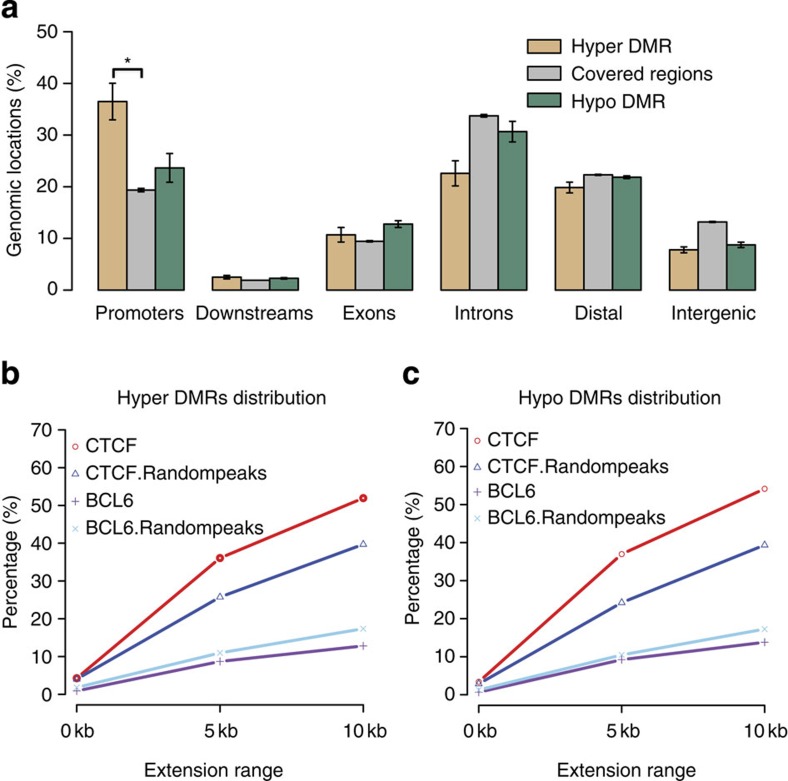
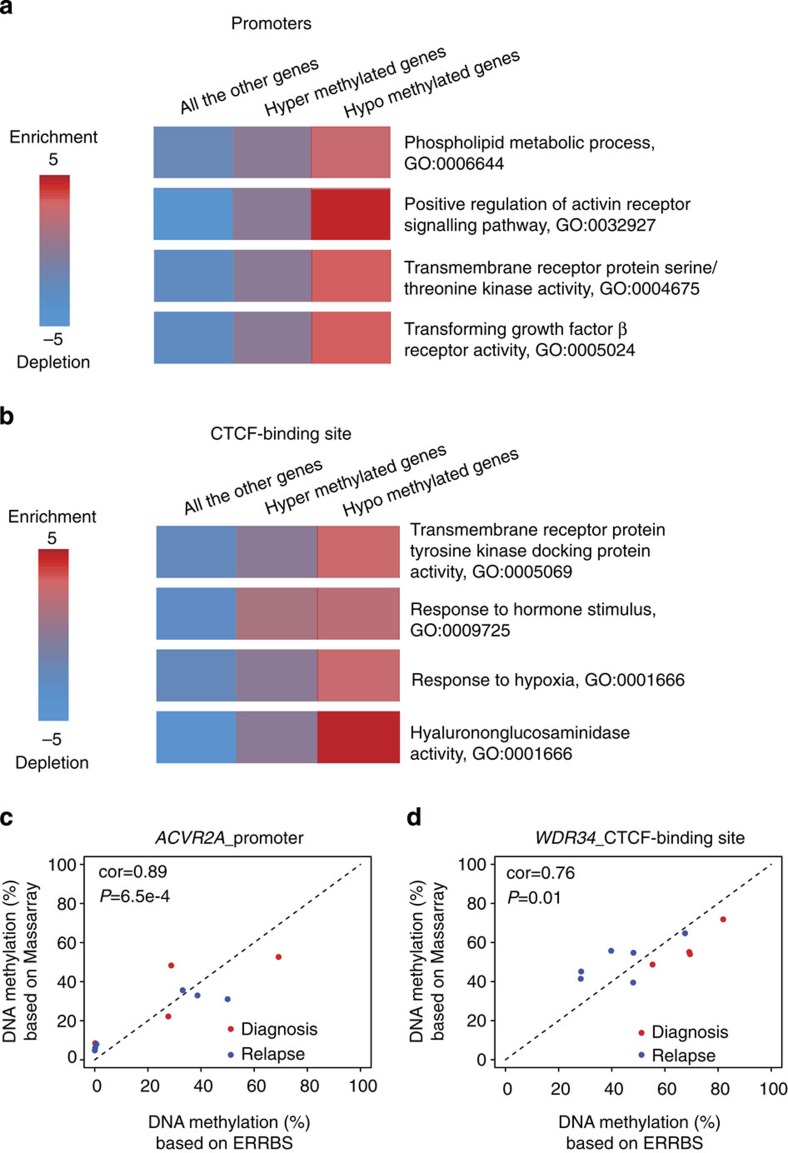
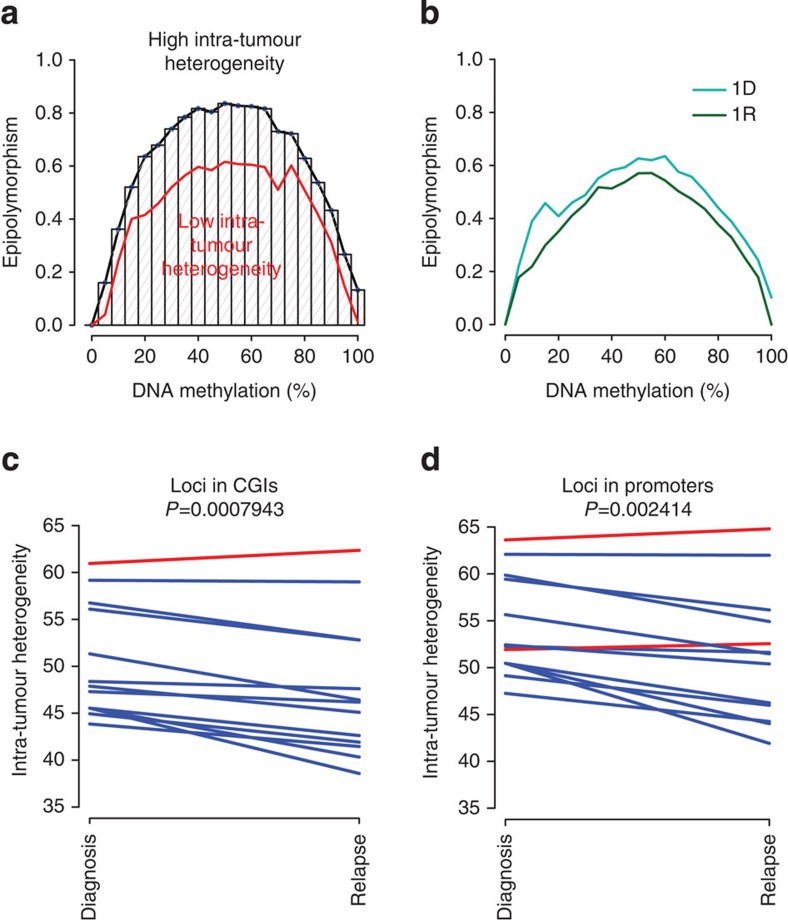
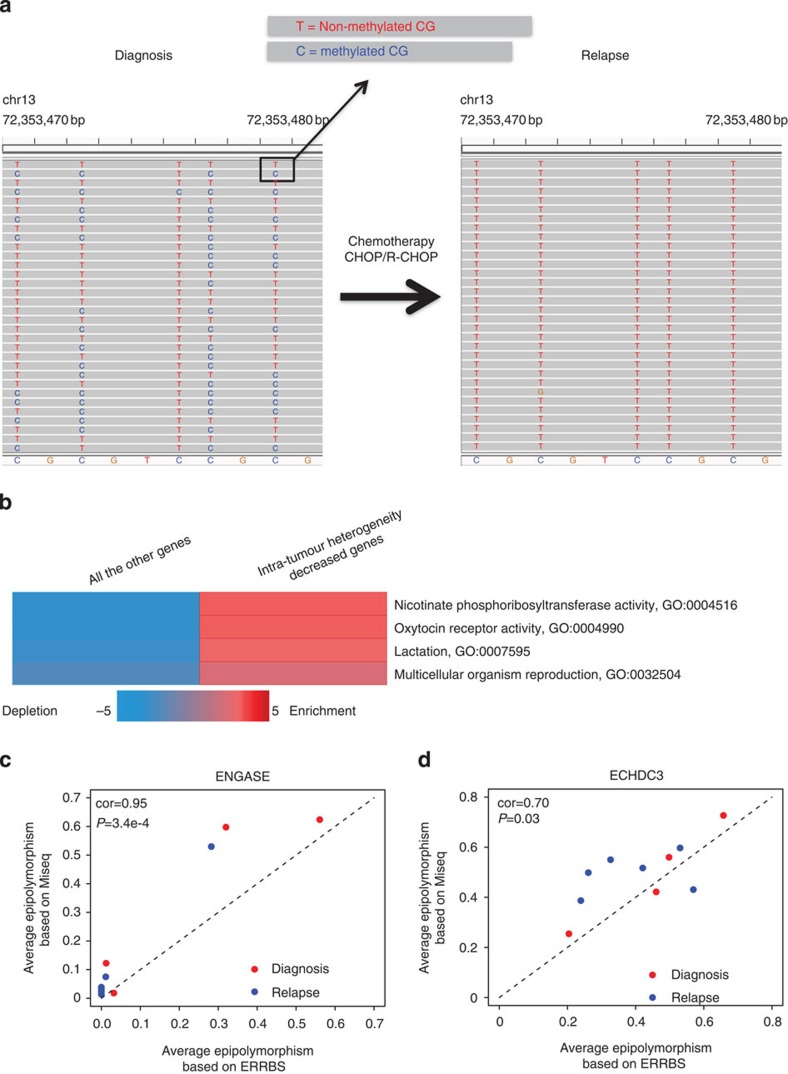
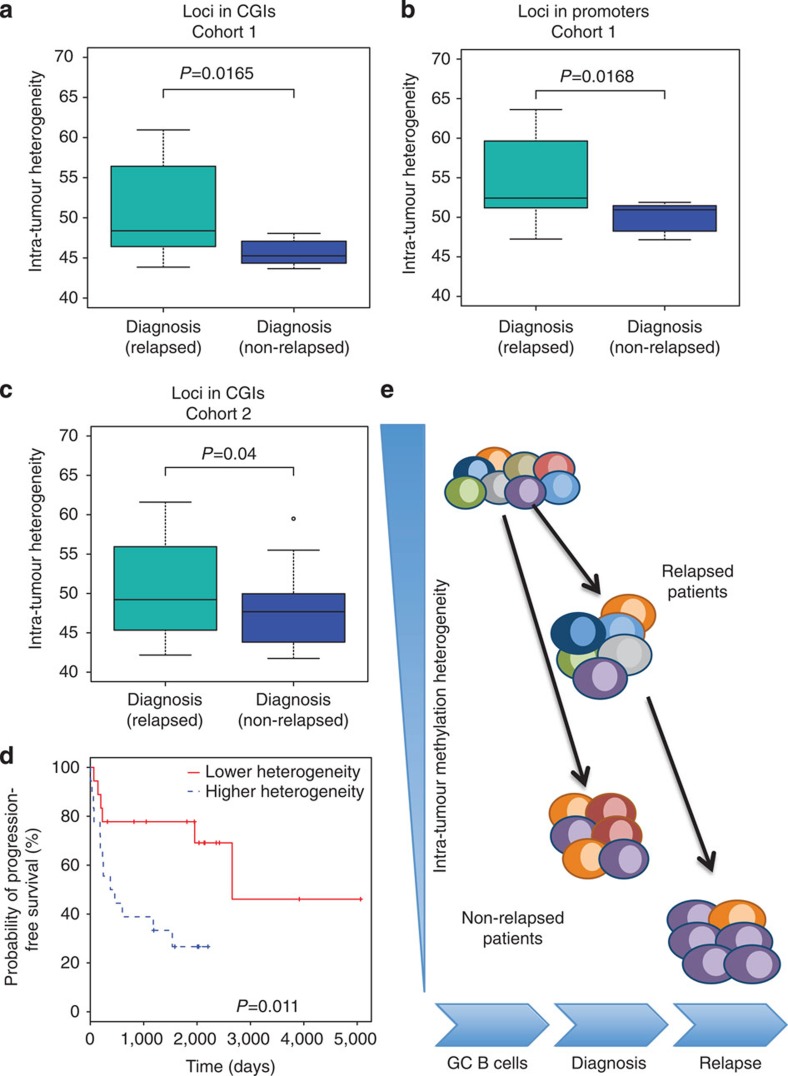
The contribution of epigenomic alterations to tumour progression and relapse is not well characterized. Here we characterize an association between disease progression and DNA methylation in diffuse large B-cell lymphoma (DLBCL). By profiling genome-wide DNA methylation at single-base pair resolution in thirteen DLBCL diagnosis-relapse sample pairs, we show that DLBCL patients exhibit heterogeneous evolution of tumour methylomes during relapse. We identify differentially methylated regulatory elements and determine a relapse-associated methylation signature converging on key pathways such as transforming growth factor-β (TGF-β) receptor activity. We also observe decreased intra-tumour methylation heterogeneity from diagnosis to relapsed tumour samples. Relapse-free patients display lower intra-tumour methylation heterogeneity at diagnosis compared with relapsed patients in an independent validation cohort. Furthermore, intra-tumour methylation heterogeneity is predictive of time to relapse. Therefore, we propose that epigenomic heterogeneity may support or drive the relapse phenotype and can be used to predict DLBCL relapse.
Figures
References
-
- Armitage J. O. & Weisenburger D. D. New approach to classifying non-Hodgkin's lymphomas: clinical features of the major histologic subtypes. J. Clin. Oncol. 16, 2780–2795 (1998). - PubMed
-
- Coiffier B. et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242 (2002). - PubMed
-
- Larouche J.-F. et al. Lymphoma recurrence 5 years or later following diffuse large B-cell lymphoma: clinical characteristics and outcome. J. Clin. Oncol. 28, 2094–2100 (2010). - PubMed
-
- Abramson J. S. & Shipp M. A. Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach. Blood 106, 1164–1174 (2005). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
